
Abstract
Aims/hypothesis
Excessive secretion of glucagon partially contributes to the development of diabetic hyperglycaemia. However, complete blocking of glucagon action will lead to adverse effects, since glucagon exerts certain beneficial effects via its receptor in many organs. We aimed to study the effects of a ‘decoy receptor’ for circulating glucagon on modulating beta cell function and glucose homeostasis in mice by over-producing the glucagon receptor (GCGR) in skeletal muscles.
Methods
We generated transgenic mice in which the expression of Gcgr is driven by the muscle specific creatine kinase (Mck) promoter, and assessed the effects of glucagon on the modulation of glucose homeostasis under conditions of extremes of glucose influx or efflux.
Results
Mck/Gcgr mice showed increased circulating levels of glucagon and insulin, resulting in an unchanged ratio of glucagon-to-insulin. The levels of hepatic glucose-6-phosphatase (G6PC) and fructose-1,6-bisphosphatase (F1,6P2ase) were significantly decreased, whereas the phosphorylation level of pancreatic cAMP-response-element-binding-protein (CREB) was significantly increased in these transgenic mice. Under basal conditions, the mice displayed normal blood glucose levels and unchanged glucose tolerance and insulin sensitivity when compared with their age-matched wild-type (WT) littermates. However, following multiple low-dose streptozotocin injections, Mck/Gcgr mice exhibited a delay in the onset of hyperglycaemia compared with the WT controls. This was associated with preserved beta cell mass and beta cell secretory capacity in response to glucose challenge.
Conclusions/interpretation
We suggest that mild and chronic hyperglucagonaemia, through a strategy involving neutralising peripheral glucagon action, provides beneficial effects on beta cell function and glucose homeostasis. Mck/Gcgr mice thus represent a novel mouse model for studying the physiological effects of glucagon.
Similar content being viewed by others

Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Introduction
Glucagon exerts its physiological functions through activation of the glucagon receptor (GCGR), a G-protein-coupled receptor, expressed in a variety of tissues including liver, kidney, brain and pancreatic beta cells [1]. Glucagon is predominant in maintaining glucose homeostasis, which represents the major counter-regulatory mechanism opposing the glucose-lowering effects of insulin [2, 3]. When hypoglycaemia occurs, glucagon stimulates hepatic glucose production by enhancing glycogenolysis and gluconeogenesis [1].
Excessive glucagon secretion contributes to the development of diabetic hyperglycaemia, due to increased hepatic glucose production either under basal or postprandial conditions [4–6]. In mice, chronic hyperglucagonaemia achieved by glucagon perfusion was shown to result in hyperglycaemia, impaired glucose tolerance and glomerular abnormalities, all symptoms that are representative of early-stage type 2 diabetes [7]. Interestingly, acute infusion of glucagon resulted in sustained hyperglycaemia only in human diabetic participants with impaired beta cell function, but not in non-diabetic participants [8]. Consistently, in insulin-withdrawn diabetic patients, the glycaemic response to hyperglucagonaemia is 5 to 15 times greater than in non-diabetic controls [5]. This suggests that, in diabetic individuals, abnormalities in the secretion of not only glucagon but also insulin, along with the alteration in the glucagon-to-insulin ratio, leads to hyperglucagonaemia [1, 9].
Ablation of glucagon action has been suggested as a potential tool for diabetes treatment [10–12]. The therapeutic strategies tested to date include the creation of Gcgr −/− mice, the use of the Gcgr antisense oligonucleotides (ASOs) or the anti-GCGR antibody. Gcgr −/− mice showed lower blood glucose levels, hyperglucagonaemia and pancreatic alpha cell hyperplasia [10]. These mice were resistant to streptozotocin (STZ)-mediated beta cell loss and hyperglycaemia [11]. Studies in humans using GCGR antagonists have shown a significant reduction in hepatic glucose production and the prevention of hyperglycaemia following glucagon infusion [12]. Consistently, the inhibition of GCGR by ASOs significantly reduced the circulating levels of triacylgycerol, NEFA and glucose in db/db mice [13]. Furthermore, the use of GCGR neutralising antibody improved glycaemic control in obese or diabetic mouse models [14, 15].
Complete blocking of glucagon action may lead to adverse effects, as glucagon exerts certain beneficial effects via its receptor in numerous organs [1]. This is exemplified by the observation that mice lacking GCGR displayed increased fetal lethality and defects in islet development and maturation [16]. Here we have generated and characterised a transgenic mouse in which the expression of Gcgr is driven by the muscle specific creatine kinase (Mck, also known as Ckm) promoter [17]. Over-production of GCGR in the skeletal muscle, where little GCGR is endogenously produced [18, 19], may produce minimal glucagon-relevant effects in the muscle. This would generate a decoy receptor for circulating glucagon to prevent potential excessive glucagon action in the peripheral tissues, while minimising the side effects that occur with total ablation of glucagon signalling. We show here that these transgenic mice exhibited increased circulating levels of glucagon and insulin, and thus the ratio of glucagon to insulin was not altered. Furthermore, the Mck/Gcgr mice showed reduced expression of genes for hepatic gluconeogenesis and glycogenolysis, but increased pancreatic phosphorylation levels of cAMP-response-element-binding-protein (CREB). Ex vivo and in vitro investigations suggested that glucagon enhances CREB phosphorylation and stimulates insulin secretion in the beta cell. We suggest that elevation of intra-islet glucagon and reduced hepatic glucagon action provide beneficial effects in glucose disposal under conditions of extremes of glucose influx or efflux.
Methods
Plasmid construction
(1) Mck/Luc: the 3.3 kb Mck promoter sequence was excised from the pEtCAT vector by digesting with BsteII, blunt-ended with Klenow and subcloned into the NheI and SmaI sites of the luciferase reporter vector pGL3-Basic. (2) CMV/Gcgr-Myc: the 1.5 kb Gcgr cDNA (Missouri S&T cDNA Resource Center) was inserted into the EcoRI and XbaI sites of pcDNA3.1Myc/His by cohesive end ligation. (3) Mck/Gcgr-Myc: Myc-tagged Gcgr cDNA was excised from CMV/Gcgr using HindIII and PmeI. The Mck/Luc construct was digested with XbaI, blunt-ended using Klenow and then digested with HindIII. The Luciferase coding sequence was replaced with Gcgr-Myc to generate the Mck/Gcgr-Myc fusion gene construct.
DNA pronuclear microinjection
The Mck/Gcgr-Myc plasmid DNA was used in microinjection for the generation of Mck/Gcgr transgenic mice (Hospital for Sick Children Transgenic Core). The transgenic mice generated are in the FVB background.
Animal care
Mice were housed under a 12 h light–dark cycle with free access to standard rodent chow and water except where noted. An intraperitoneal glucose tolerance test (IPGTT), insulin tolerance test (ITT) and glucagon tolerance test (GcgTT) were performed as described previously [20]. Insulin-induced hypoglycaemia was performed in randomly fed mice by intraperitoneal insulin injection (2.0 U/kg) and blood glucose levels were monitored at the indicated times. Diabetic hyperglycaemia was induced by multiple low-dose STZ injection as described previously [20]. Metabolic cages were used in some groups of mice before termination of the experiments. All procedures complied with guidelines approved by the Canadian Council of Animal Care and institutional animal care committee.
Genotyping
A tail sample (1 mm) was cut for genotyping with a pair of primers that amplify the inserted transgene Gcgr-Myc cDNA (electronic supplementary material [ESM] Fig. 1).
Plasmid transfection
The L6 muscle cell line and hamster kidney fibroblast cell line BHK were maintained in culture [21, 22]. Lipofectamine 2000 (Invitrogen, Burlington, ON, Canada) was used for plasmid transfections.
Luciferase (LUC) assay
Cells were transfected with the control pGL3-Basic or Mck/Luc plasmids. Forty-eight hours after the transfection, cell lysates were used for LUC reporter analysis [22].
Ligand-receptor binding assay
Cell plasma membranes were prepared as described previously [23] from L6 muscle cells transfected with or without CMV/Gcgr. Competitive binding studies were performed essentially as described in Unson et al. [24] in 20 μg of membrane proteins with 125I-labelled glucagon (20,000 cpm, Perkin Elmer), in the presence of indicated cold glucagon.
2-Deoxy-d-glucose uptake
Glucose transport was determined in transfected or non-transfected L6 cells grown in a 24-well plate, treated with or without insulin (100 nmol/l, 20 min) after serum deprivation as described previously [23].
mRNA analysis
Total RNA was used for the generation of cDNA for assessing Gcgr expression in mouse tissues as previously described [17]. SYBR green-based real-time PCR was used to evaluate hepatic gene expression of glucose-6-phosphatase (G6pc) and fructose-1,6-bisphosphatase (F1,6P2ase, also known as Fbp1). Primer sequences are listed in ESM Table 1.
Western blotting
Western blot analysis was performed using mouse monoclonal anti-MYC tag antibody (Upstate, MA, USA, 1:1,000), or rabbit monoclonal anti-CREB antibody (Cell Signaling, 1;1,000), detected with HRP-conjugated relevant secondary antibodies (1:5,000, Jackson Lab, ME, USA) and visualised by Enhanced Chemiluminescence (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). For phosphorylation assay, INS-1 cells [25] were serum-deprived for 16 h and incubated with glucagon at indicated concentrations, or insulin (100 nmol/l) in the presence or absence of inhibitors for an additional 15 min.
Southern blotting
A probe was generated by digestion of Mck/Gcgr with ApaI and BamHI to obtain the 449 bp Gcgr fragment, and then 32P-labelled using a random prime labelling system (Amersham, Chalfont St Giles, UK). Genomic DNA was digested overnight at 37°C with ApaI and transferred onto a nitrocellulose membrane, hybridised with the probe (2 × 106 cpm/ml) and the autoradiograph was developed on an X-ray film [23].
Radioimmunoassay and enzyme-linked immunosorbent assay
Serum glucagon and C-peptide, insulin or glucagon-like peptide-1 (GLP-1) concentrations were determined [20] using corresponding RIA kits (Linco, St Charles, MO, USA). Plasma adrenalin (epinephrine) levels were determined using an ELISA kit (IBL, Hamburg, Germany).
Immunohistochemistry and islet cell mass analysis
Tissue isolation, processing and paraffin embedding were conducted as described previously [20, 26]. Pancreatic sections (5 μm) were used for insulin and glucagon staining and detected by chromogenic methods as described previously [20, 26]. The beta and alpha cell mass was determined as the product of the total cross-sectional beta or alpha cell areas over total tissue area and the weight of the pancreas before fixation as described previously [20, 26]. For muscle tissue staining, anti-MYC mouse IgG (1:500) was used followed by Alexafluor555 anti-mouse (Molecular Probes, OR, USA, 1:500).
Mck driven Gcgr expression
The transgene vector Mck/Gcgr was tested in vitro prior to the pronuclear microinjection. Immunocytochemistry was performed in myotubes co-transfected with Mck/Gcgr and GFP cDNAs, using the anti-MYC antibody (ESM Fig. 2a). Western blotting detected the Mck/Gcgr expression only in L6 muscle cells but not in Cos-7 control cells (ESM Fig. 2b), indicating the muscle specific nature of the Mck promoter.
The 125I-labelled glucagon binding in the membranes from Gcgr-transfected muscle cells, in the presence of cold glucagon, displayed a typical homologous competitive binding pattern (ESM Fig. 2c), suggesting specific GCGR–glucagon binding. Little binding was observed either in mock-transfected or non-transfected cell membranes, indicating low endogenous GCGR levels in muscle cells, consistent with previous reports that muscle cells produce very little, if no, GCGR [18, 19].
A glucose transport assay was performed and we found that ectopic GCGR production in the muscle cells did not alter either basal or insulin-stimulated glucose uptake (ESM Fig. 2d).
The generation of transgenic Mck/Gcgr mice
Pronuclear injections with Mck/Gcgr-Myc produced 14 pups. Five Mck/Gcgr + founders (denoted A, B, C, D and E/F, ESM Fig. 3) were bred into the CD1 background to produce F1 and subsequent generations. Genomic transgene integrations in these founders were determined by Southern blotting (ESM Fig. 4). Three (lines A, D and E/F) of the five lines were found to carry the transgene.
A Gcgr mRNA tissue distribution assay showed that Gcgr expression was predominantly in the liver and kidney, with lower expression in other tissues, including the pancreas and brain, but not in muscles (ESM Fig. 5a). Substantially increased Gcgr mRNA levels were present in the muscles of the transgenic mice, while no obvious changes were observed in other tissues (ESM Fig. 5a). GCGR protein was expressed in muscle cells of three transgenic lines as confirmed by western blotting and immunohistochemistry (ESM Fig. 5b, c). We selected the founder F, based on the expression abundance of the transgene, for all subsequent experiments.
Statistical analysis
All data were presented as mean ± SEM. Statistical analysis was performed using unpaired two-tailed Student’s t test or ANOVA with Bonferroni’s multiple comparisons post hoc test where appropriate. Data analysis was conducted using Graph Pad software.
Results
Phenotypic characterisation of the Mck/Gcgr mice
Increases in body weight were observed in the transgenic mice of both sexes (Fig. 1a, b). In the F1 generation, the increased body weight was shown to be associated with increased food intake in the male mice but not obviously in the female mice (ESM Fig. 6a, b). No difference was found in fasting or fed blood glucose levels between the Mck/Gcgr mice and the wild-type (WT) littermates (ESM Fig. 6c, d). IPGTT, ITT and GcgTT showed no difference between the transgenic mice and the WT littermates (ESM Fig. 6e–g), suggesting that the Mck/Gcgr mice had minimal change in glucose homeostasis under basal conditions.

Phenotypic characterisation of the Mck/Gcgr mice. Measurements of the body weight in female (a) and male (b) Mck/Gcgr mice (black bars) and their age-matched WT littermates (white bars) from 4 to 16 weeks of age. c Detection of circulating levels of glucagon, C-peptide (d) and GLP-1 (e) in the Mck/Gcgr mice and their age-matched WT littermates by RIA. f Pancreatic sections from the Mck/Gcgr mice and their age-matched WT littermates (16 weeks of age) were stained for insulin (red) and glucagon (dark brown). Scale bars 30 μm. Analysis of beta cell mass (g) and alpha cell mass (h) are shown. i The graph shows islet number (%) vs islet size measured; white squares, WT; black diamonds, Mck/Gcgr; the inset (bar graph) shows the measurement of the number of islets of very small size (0.1–0.2 mm2). Quantification of hepatic mRNA expression of G6PC (j) and F1,6P2ase (k) by quantitative PCR (qPCR) in the Mck/Gcgr mice and their age-matched WT littermates. Data are mean ± SEM, *p < 0.05, n = 5–10
A RIA showed that serum glucagon levels were increased in the Mck/Gcgr mice by 42% compared with the WT littermates (Fig. 1c, p < 0.05). The serum C-peptide levels in the transgenic mice were increased by 60% compared with the WT littermates (Fig. 1d, p < 0.05). However, serum GLP-1 levels were not changed in Mck/Gcgr mice (Fig. 1e, p > 0.05). These results suggest that ectopic GCGR production in muscles leads to increases in circulating glucagon levels as well as insulin, hence maintaining an appropriate glucagon-to-insulin ratio.
The islet architecture of the transgenic mice was not altered (Fig. 1f). Islet cell mass analysis showed no statistical difference in beta cell mass or alpha cell mass between Mck/Gcgr mice and the WT littermates (Fig. 1g, h, p > 0.05). However, although not statistically significant, the numbers of small islets (i.e., 0.1–0.2 mm2) in the transgenic mice tended to be increased by 30% (Fig. 1i).
Although the transgenic animals showed mild hyperglucagonaemia, their hepatic G6pc and F1,6p2ase mRNA levels were found to be significantly reduced (Fig. 1j, k), suggesting reduced expression of genes involved in hepatic gluconeogenesis and glycogenolysis in the transgenic mice.
Greater glucagon secretory response during hypoglycaemia in Mck/Gcgr mice
We thought to examine whether the ectopic production of Gcgr in muscle cells would alter the glucagon counter-regulatory response in response to hypoglycaemia. Administration of insulin (i.p., 2 U/kg) produced profound hypoglycaemia in the random-fed mice (Fig. 2a). Analysis of counter-regulatory response to hypoglycaemia showed a significantly greater increase in circulating glucagon levels in the Mck/Gcgr mice compared with the WT control mice (Fig. 2b, p < 0.05), suggesting that, in order to maintain appropriate glucose levels during a period of hypoglycaemia, Mck/Gcgr mice produced twofold more glucagon than WT mice. In contrast, the secretory response of adrenaline to the profound hypoglycaemia in both groups of mice were not statistically different (Fig. 2c, p > 0.05).

Greater glucagon secretory response during hypoglycaemia in Mck/Gcgr mice. a Blood glucose levels were measured during an insulin-induced (2 U/kg) hypoglycaemia in Mck/Gcgr mice (black squares) and WT control mice (white diamonds). b 40 min after insulin injection (2 U/kg), mice were killed and analysed for serum glucagon levels by RIA, or c plasma adrenaline levels by ELISA
Protective effect of muscle GCGR production in the STZ-induced diabetes mouse model
To investigate whether ectopic production of GCGR in muscle cells would provide a beneficial effect on glucose disposal following a challenge in the mice, we investigated the effects of hyperglycaemia in the Mck/Gcgr mice using a multiple-low-dose STZ-induced diabetes (MDSD) strategy. As shown, STZ injections induced severe hyperglycaemia in the WT littermates, while blood glucose levels in the Mck/Gcgr mice were remarkably lower at each indicated time point (Fig. 3a). When the glycaemic levels during the feeding course were expressed as the AUC, the difference between the two groups was statistically significant (Fig. 3b, p < 0.05). IPGTT performed 36 days after STZ injections showed lower glycaemic excursion in the Mck/Gcgr mice (Fig. 3c, d, p < 0.05), suggesting improved glucose tolerance in the transgenic mice under the MDSD hyperglycaemic conditions.

Protective effect of muscle GCGR production in the STZ-induced diabetes mouse model. a Mck/Gcgr mice and their age-matched WT littermates at the age of 14 weeks were i.p. injected with STZ (50 mg/kg, for four consecutive days). Blood glucose levels were measured at indicated time point during the feeding course; white diamonds, WT; black squares, Mck/Gcgr. b The AUCs are shown for the experiment at (a). c An IPGTT was performed at day 36 after the STZ injections; white diamonds, WT; black squares, Mck/Gcgr. d The AUCs for the glycaemic curves are shown. e Representative pancreatic section stained for insulin (red) and glucagon (dark brown) from the normal control background CD1 mice (Ctrl), or the WT, or the Mck/Gcgr mice, following STZ injections. Analysis of the beta cell mass (f) and alpha cell mass (g) from these mice are shown. Data are mean ± SEM. *p < 0.05, n > 5
We then considered whether the improved glucose tolerance in the Mck/Gcgr mice following STZ injection was due to enhanced beta cell mass or function. As shown (Fig. 3e), STZ injections destroyed the islet architecture and caused a remarkable loss of beta cells in the WT animals. In contrast, Mck/Gcgr mice showed close to normal islet architecture and preserved beta cell mass (Fig. 3e, f). The alpha cell mass appeared not to be significantly changed in either group following STZ injections (Fig. 3g).
To evaluate the beta cell secretory function, we performed an indirect assessment of pancreatic insulin release in which plasma insulin concentrations were determined by RIA during an IPGTT (Fig. 4a). As shown, 15 min after glucose administration, STZ-WT mice showed almost no insulin response to the glucose challenge. In contrast, the insulin response was better in the transgenic than the WT mice, consistent with their well preserved beta cell mass (Fig. 3e, f). A glucagon RIA showed that glucose-induced suppression of glucagon secretion, as seen in normal control mice, was diminished in the STZ-WT mice but was maintained in the STZ-Mck/Gcgr mice (Fig. 4b).

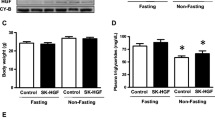
Effects of STZ on glucose-induced insulin and glucagon release in the Mck/Gcgr mice. Measurements of plasma insulin concentrations (a) and glucagon concentrations (b) during an IPGTT in the normal control background CD1 mice (Ctrl), or the WT, or Mck/Gcgr mice prior to and 36 days after STZ injections; for (a) and (b), white bars, 0 min; black bars, 15 min. c Metabolic studies were conducted using a metabolism cage. d The loss of body weight was compared at the end of the feeding course; for (c) and (d) white bars, WT, black bars Mck/Gcgr. Data are means ± SEM *p < 0.05, **p < 0.01, † p < 0.05 (Mck/Gcgr vs WT), n = 5
The STZ-Mck/Gcgr mice showed improved polyphagia, polyuria and polydipsia and reduced body weight loss, compared with the STZ-WT mice (Fig. 4c, d).
Increased pancreatic CREB phosphorylation in the transgenic mice
CREB coordinates the actions of glucose, GLP-1 and glucagon. It is crucial in maintaining beta cell survival and glucose homeostasis [27, 28]. We found that, in the Mck/Gcgr mice, pancreatic phosphorylated CREB (pCREB) levels were significantly higher than those of WT mice (Fig. 5a, b; p < 0.05).

Increased pancreatic CREB phosphorylation in the transgenic mice. a A representative western blot shows the detection of phosphorylated CREB (pCREB) in pancreatic tissue from the WT or Mck/Gcgr mice, and (b) the quantitative analysis of panel (a). c Detection of pCREB in INS-1 cells treated with glucagon at indicated concentrations in the absence or presence of H89 or nifidipine (Nif) and (d) the quantitative analysis of panel (c). e Detection of insulin secretion by RIA in INS-1 cells treated with glucagon at indicated concentrations, in the presence of low (LG; 2.8 mmol/l) or high glucose (HG; 16.7 mmol/l). f Western blot analysis of pCREB in INS-1 cells treated with 100 nmol/l insulin for indicated times. Data are means ± SEM, *p < 0.05, **p < 0.01, n = 3–5
Glucagon stimulates pCREB levels and insulin secretion in pancreatic beta cells
The transgenic mice displayed increased circulating glucagon and insulin, but not GLP-1 levels (Fig. 1b–d); we wondered whether elevated intra-islet glucagon (or insulin) is responsible for increased pancreatic pCREB in Mck/Gcgr mice. In vitro investigations were performed in the INS-1 cell line to address this question. As shown, glucagon dose-dependently stimulated CREB phosphorylation in this beta cell line, which was blocked by the protein kinase A (PKA) inhibitor H-89, or by the calcium channel blocker nifedipine (Fig. 5c, d). Furthermore, glucagon was also shown to dose-dependently increase insulin secretion in this cell line (Fig. 5e). Finally, insulin was shown to increase pCREB levels in INS-1 cells as well (Fig. 5f).
Discussion
We have generated transgenic mice that ectopically express Gcgr in the skeletal muscles. Since it is unlikely that the binding of glucagon to the cloned GCGR in muscles induces a predominant physiological effect, neutralising excessive glucagon by muscle cells would reduce the action of glucagon, particularly under hyperglucagonaemic conditions. This novel mouse model allowed us to test the feasibility of neutralising circulating glucagon and the impact this had on glucose homeostasis.
Mck/Gcgr transgenic mice displayed normal fasting and random-fed glucose levels, glucose tolerance and insulin sensitivity compared with their age-matched littermates. This differs from phenotypic characterisation of Gcgr −/− mice, which showed lower blood glucose levels, normal insulin levels and improved glucose tolerance [10].
Mck/Gcgr mice displayed increased circulating glucagon levels, presumably representing a compensatory mechanism in response to the neutralisation of glucagon in the circulation. Glucagon is a primary regulator of hepatic glucose production. Prior to its appearance in the circulation, a considerable amount of glucagon is trapped by the liver. Glucagon kinetics studies have revealed that it is rapidly cleared from the plasma, primarily by liver and kidneys [29]. Interestingly, glucagon trapped in the liver was saturable and reversible, since a sequential injection of 125I-labelled glucagon and excess native glucagon resulted in a remarkable displacement of 125I-labelled glucagon from the liver back into the plasma [29]. In the Mck/Gcgr mice, the ectopic production of GCGR was at comparable levels to that of the liver and kidneys. Given the much greater muscle mass compared with that of liver and kidneys, the ectopic production of GCGR in the muscle thus provides effective ‘decoy receptors’. However, unlike the robust hyperglucagonaemia observed in the Gcgr −/− mice [10] or the mice receiving therapeutic intervention by GCGR antagonistic strategies [13, 15], Mck/Gcgr mice displayed a relatively mild increase in glucagon levels. Furthermore, elevated glucagon levels were associated with increased insulin levels in Mck/Gcgr mice, allowing the maintenance of an appropriate glucagon-to-insulin ratio, which is critical in maintaining glycaemic stability, particularly in extremes of glucose influx or efflux [6, 30].
While the glycaemic recovery from an insulin-induced hypoglycaemia was found to be no different between the transgenic mice and the WT littermates, the transgenic mice displayed a greater glucagon counter-regulatory response. This greater glucagon secretory response in our Mck/Gcgr mice may represent an important mechanism to compensate the glucagon trapping in the muscle in Mck/Gcgr mice. Interestingly, the adrenaline secretory response to the profound hypoglycaemia in both groups of mice was not statistically different, consistent with the notion that adrenaline, as the third physiological defence after insulin and glucagon, only becomes critical when the glucagon counter-regulatory response is impaired [31].
In the Mck/Gcgr mice, although the number of small islets tends to be increased, islet beta and alpha cell masses in these mice are unchanged. This suggests that the elevation of glucagon levels observed in these mice is a consequence of increased alpha cell secretory capacity. It is conceivable that elevated intra-islet glucagon could stimulate beta cell secretion, leading to increased circulating insulin levels in the Mck/Gcgr mice.
In the MDSD type 1 diabetes model, Mck/Gcgr mice were resistant to the development of hyperglycaemia and displayed much improved diabetic symptoms, which is consistent with the findings in the Gcgr −/− mice [11]. Detailed mechanisms underlying the resistance to STZ in these two mouse models remain largely unknown, although increased circulating GLP-1 has been proposed to partially explain STZ resistance in the Gcgr −/− mice [10]. Our Mck/Gcgr mice displayed unchanged circulating GLP-1 levels, increased glucagon levels, and increased levels of insulin, while the latter was shown to be reduced in the Gcgr −/− mice [10]. It is conceivable that the intra-islet glucagon concentration is much higher than its concentration in the circulation in the Mck/Gcgr mice. We therefore speculate that elevated intra-islet glucagon action exerts a paracrine effect on beta cells, rendering them more competent. This notion is supported by the evidence that endogenous glucagon promotes glucose competence of insulin release in human islets [32]. Pancreas perfusion studies using specific receptor antagonists demonstrated that glucagon stimulates insulin release mainly through GCGR, but not GLP-1 receptors on islet beta cells [33]. The critical role of glucagon in maintaining beta cell function is also supported by the observations in Gcgr −/− mice, which displayed impaired beta cell function due to the lack of glucagon action [34]. Consistently, this perception is further supported by another study showing that beta cell specific over-production of GCGR enhanced glucose-competent insulin secretion and increased beta cell mass [35].
After STZ injection, the WT mice had severely depleted beta cell mass, whereas the loss of beta cell mass in the Mck/Gcgr mice was considerably less, accompanied by a delay in the onset of hyperglycaemia in these transgenic mice. It is conceivable that the preserved beta cell mass (Fig. 3e) and beta cell secretory capacity (Fig. 4a) rendered the Mck/Gcgr mice able to maintain glucose-induced insulin secretion and to suppress glucagon secretion (Fig. 4b), under the beta cell injuring conditions (STZ). This is also consistent with recent findings that intra-islet insulin is a major mechanism underlying glucose-mediated suppression of glucagon secretion in alpha cells [36, 37].
CREB is crucial for beta cell function and survival [28]. In insulin-secreting cell lines and isolated rodent islets, Dalle et al. [38] demonstrated that glucagon could stimulate CREB phosphorylation via a PKA/MEK/ERK dependent signalling pathway. The hyperglucagonaemic Mck/Gcgr mice displayed significantly increased pancreatic pCREB, suggesting that glucagon-stimulated CREB activation underlies enhanced beta cell competency in these transgenic mice. In the INS-1 cell line, we showed that glucagon stimulates CREB activation, possibly via PKA and calcium signalling pathways. Interestingly, insulin also enhanced CREB phosphorylation in a PI3-K dependent fashion (data not shown). These observations collectively suggest that both paracrine (glucagon) and autocrine (insulin) action within the islets promote beta cell competency in Mck/Gcgr mice, particularly after either beta cell injury or excessive metabolic stress conditions.
Enhanced insulin secretion in Mck/Gcgr mice is presumably an additional mechanism that renders beta cells more competent to STZ-induced beta cell injury and apoptosis, considering that autocrine insulin action is pivotal in maintaining beta cell secretion and function [39–41]. Insulin prevents not only beta cell death during excessive nutrition-induced oxidative stress, but also cytokine-induced beta cell death [40, 42]. The beneficial effect of insulin on pancreatic beta cells is mediated by phosphoinositide 3-kinase (PI3-K)/protein kinase B (AKT) signalling [43, 44], since a reduced beta cell autocrine PI3-K/AKT pathway was shown to contribute to beta cell apoptosis in the progression of diabetic hyperglycaemia in a rodent model [45]. We suggest that autocrine insulin action within the islets could be mediated by the insulin–PI3-K–CREB signalling pathway [46].
Glucagon stimulates glucose production in liver mostly through enhancing glycogenolysis, with little effect on gluconeogenesis [47]. Following hypoglycaemia, decreased insulin signalling and increased glucagon action enhance PKA-dependent glycogen phosphorylase activity while suppressing glycogen synthase activity, leading to glycogen breakdown [47], a process involved in the activation of AMP-activated protein kinase (AMPK) [48]. Of note, insulin exerts both portal and peripheral effects to suppress hepatic glucose production, counteracting the effect of glucagon. In Mck/Gcgr mice, the increased circulating glucagon level, which is still within the physiological range, is associated with increased plasma insulin concentration. This may allow insulin to exert high control strength over the rate of hepatic glucose output, thereby maintaining glucose homeostasis.
We anticipated that, in the Mck/Gcgr mice, glucagon action in the liver is reduced as a result of the titration of glucagon by the decoy receptor in the muscle. Reduced expression of genes involved in hepatic gluconeogenesis and glycogenolysis in Mck/Gcgr mice is one of the explanations for why Mck/Gcgr mice have normal glucose levels despite of the presence of increased circulating glucagon. It should be pointed out that glucagon injection in the transgenic mice showed a similar effect on plasma glucose levels when compared with the WT littermates (ESM Fig. 6). This could be, at least in part, caused by the increased insulin secretion and enhanced insulin action in the liver (Fig. 6). Suppressive effects of insulin on hepatic glucose production have been well documented. Insulin signalling in the liver is critical for the proper maintenance of metabolic homeostasis [49]. Particularly, in the absence of insulin signalling, insulin cannot suppress hepatic glucose production in liver-specific insulin receptor knockout (LIRKO) mice [50]. The Mck/Gcgr mice have both elevated glucagon and insulin, thus maintaining an appropriate ratio of glucagon to insulin, which represents an important mechanism for maintaining glycaemic stability, particularly in extremes of glucose influx or efflux [9] (Fig. 6).

A model shows mechanisms underlying the beneficial effects of ectopic production of GCGR in skeletal muscles. (1) Expression of cloned Mck/Gcgr in muscles creates a ‘decoy receptor’ for circulating glucagon. (2) Elevation of glucagon increases pancreatic CREB phosphorylation and glucose-competent beta cell secretion. (3) Elevation of insulin secretion increases the suppressive effect of insulin on hepatic glucose production by reducing G6PC and F1,6P2ase levels. (4) Elevation of glucagon does not increase its action in the liver because of simultaneously elevated insulin that maintains an appropriate ratio of glucagon to insulin. The beta cell autocrine trophic effects and its paracrine suppressive effects on alpha cells are not shown
In conclusion, we have generated a novel transgenic mouse model with muscle specific expression of Gcgr. The transgenic mice display increased circulating levels of both glucagon and insulin and hence maintain an appropriate ratio of glucagon to insulin, which appears important in maintaining glucose homeostasis. While the Mck/Gcgr mice display minimal change in glucose homeostasis under basal conditions, they are resistant to STZ-induced hyperglycaemia. We suggest that the mechanism underlying the protection in these mice to drug-induced beta cell injury is, at least in part, due to enhanced intra-islet action of both glucagon and insulin on beta cell function. The Mck/Gcgr mouse may serve as a useful model for studying glucagon physiology.
Abbreviations
- AKT:
-
Protein kinase B
- ASO:
-
Antisense oligonucleotide
- CREB:
-
cAMP-response-element-binding-protein
- F1,6P2ase:
-
Fructose-1,6-biphosphatase
- GcgTT:
-
Glucagon tolerance test
- GLP-1:
-
Glucagon-like peptide-1
- G6PC:
-
Glucose-6-phosphatase
- GCGR:
-
Glucagon receptor
- IPGTT:
-
Intraperitoneal glucose tolerance test
- ITT:
-
Insulin tolerance test
- LUC:
-
Luciferase
- MCK:
-
Muscle specific creatine kinase
- MDSD:
-
Multiple-low-dose STZ-induced diabetes
- MYC:
-
Protein encoded by myelocytomatosis oncogene
- PI3-K:
-
Phosphoinositide 3-kinase
- PKA:
-
Protein kinase A
- STZ:
-
Streptozotocin
- WT:
-
Wild-type
References
Bansal P, Wang Q (2008) Insulin as a physiological modulator of glucagon secretion. Am J Physiol Endocrinol Metab 295:E751–E761
Jiang G, Zhang BB (2003) Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab 284:E671–E678
Gromada J, Franklin I, Wollheim CB (2007) Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev 28:84–116
Gastaldelli A, Baldi S, Pettiti M et al (2000) Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes 49:1367–1373
Sherwin RS, Fisher M, Hendler R, Felig P (1976) Hyperglucagonemia and blood glucose regulation in normal, obese and diabetic subjects. N Engl J Med 294:455–461
Unger RH (1978) Role of glucagon in the pathogenesis of diabetes: the status of the controversy. Metabolism 27:1691–1709
Li XC, Liao TD, Zhuo JL (2008) Long-term hyperglucagonaemia induces early metabolic and renal phenotypes of type 2 diabetes in mice. Clin Sci (Lond) 114:591–601
Rizza R, Verdonk C, Miles J, Service FJ, Gerich J (1979) Effect of intermittent endogenous hyperglucagonemia on glucose homeostasis in normal and diabetic man. J Clin Invest 63:1119–1123
Unger RH, Orci L (2010) Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci U S A 107:16009–16012
Gelling RW, Du XQ, Dichmann DS et al (2003) Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A 100:1438–1443
Conarello SL, Jiang G, Mu J et al (2007) Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 50:142–150
Petersen KF, Sullivan JT (2001) Effects of a novel glucagon receptor antagonist (Bay 27-9955) on glucagon-stimulated glucose production in humans. Diabetologia 44:2018–2024
Liang Y, Osborne MC, Monia BP et al (2004) Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 53:410–417
Gu W, Yan H, Winters KA et al (2009) Long-term inhibition of the glucagon receptor with a monoclonal antibody in mice causes sustained improvement in glycemic control, with reversible alpha-cell hyperplasia and hyperglucagonemia. J Pharmacol Exp Ther 331:871–881
Yan H, Gu W, Yang J et al (2009) Fully human monoclonal antibodies antagonizing the glucagon receptor improve glucose homeostasis in mice and monkeys. J Pharmacol Exp Ther 329:102–111
Vuguin PM, Kedees MH, Cui L et al (2006) Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology 147:3995–4006
Schertzer JD, Antonescu CN, Bilan PJ et al (2009) A transgenic mouse model to study glucose transporter 4myc regulation in skeletal muscle. Endocrinology 150:1935–1940
Hansen LH, Abrahamsen N, Nishimura E (1995) Glucagon receptor mRNA distribution in rat tissues. Peptides 16:1163–1166
Burcelin R, Li J, Charron MJ (1995) Cloning and sequence analysis of the murine glucagon receptor-encoding gene. Gene 164:305–310
Soltani N, Kumar M, Glinka Y, Prud'homme GJ, Wang Q (2007) In vivo expression of GLP-1/IgG-Fc fusion protein enhances beta-cell mass and protects against streptozotocin-induced diabetes. Gene Ther 14:981–988
Wang Q, Khayat Z, Kishi K, Ebina Y, Klip A (1998) GLUT4 translocation by insulin in intact muscle cells: detection by a fast and quantitative assay. FEBS Lett 427:193–197
Jin T, Drucker DJ (1995) The proglucagon gene upstream enhancer contains positive and negative domains important for tissue-specific proglucagon gene transcription. Mol Endocrinol 9:1306–1320
Wang Q, Somwar R, Bilan PJ et al (1999) Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol Cell Biol 19:4008–4018
Unson CG, Cypess AM, Kim HN et al (1995) Characterization of deletion and truncation mutants of the rat glucagon receptor. Seven transmembrane segments are necessary for receptor transport to the plasma membrane and glucagon binding. J Biol Chem 270:27720–27727
Zhang N, Kumar M, Xu G et al (2006) Activin receptor-like kinase 7 induces apoptosis of pancreatic beta cells and beta cell lines. Diabetologia 49:506–518
Wang Q, Brubaker PL (2002) Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia 45:1263–1273
Kim SJ, Nian C, Widenmaier S, McIntosh CH (2008) Glucose-dependent insulinotropic polypeptide-mediated up-regulation of beta-cell antiapoptotic Bcl-2 gene expression is coordinated by cyclic AMP (cAMP) response element binding protein (CREB) and cAMP-responsive CREB coactivator 2. Mol Cell Biol 28:1644–1656
Jhala US, Canettieri G, Screaton RA et al (2003) cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev 17:1575–1580
Balage M, Grizard J (1986) Relationship between plasma glucagon disappearance and tissue uptake in rats. Reprod Nutr Dev 26:31–38
Kuwajima M, Golden S, Katz J, Unger RH, Foster DW, McGarry JD (1986) Active hepatic glycogen synthesis from gluconeogenic precursors despite high tissue levels of fructose 2,6-bisphosphate. J Biol Chem 261:2632–2637
Cryer PE (2008) The barrier of hypoglycemia in diabetes. Diabetes 57:3169–3176
Huypens P, Ling Z, Pipeleers D, Schuit F (2000) Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia 43:1012–1019
Kawai K, Yokota C, Ohashi S, Watanabe Y, Yamashita K (1995) Evidence that glucagon stimulates insulin secretion through its own receptor in rats. Diabetologia 38:274–276
Sorensen H, Winzell MS, Brand CL et al (2006) Glucagon receptor knockout mice display increased insulin sensitivity and impaired beta-cell function. Diabetes 55:3463–3469
Gelling RW, Vuguin PM, Du XQ et al (2009) Pancreatic beta-cell overexpression of the glucagon receptor gene results in enhanced beta-cell function and mass. Am J Physiol Endocrinol Metab 297:E695–E707
Xu E, Kumar M, Zhang Y et al (2006) Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab 3:47–58
Tsuchiyama N, Takamura T, Ando H et al (2007) Possible role of alpha-cell insulin resistance in exaggerated glucagon responses to arginine in type 2 diabetes. Diabetes Care 30:2583–2587
Dalle S, Longuet C, Costes S et al (2004) Glucagon promotes cAMP-response element-binding protein phosphorylation via activation of ERK1/2 in MIN6 cell line and isolated islets of Langerhans. J Biol Chem 279:20345–20355
Aspinwall CA, Lakey JR, Kennedy RT (1999) Insulin-stimulated insulin secretion in single pancreatic beta cells. J Biol Chem 274:6360–6365
Wang Q, Jin T (2009) The role of insulin signaling in the development of beta-cell dysfunction and diabetes. Islets 1:95–101
Johnson JD, Bernal-Mizrachi E, Alejandro EU et al (2006) Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc Natl Acad Sci U S A 103:19575–19580
Paraskevas S, Aikin R, Maysinger D et al (2001) Modulation of JNK and p38 stress activated protein kinases in isolated islets of Langerhans: insulin as an autocrine survival signal. Ann Surg 233:124–133
Jetton TL, Lausier J, LaRock K et al (2005) Mechanisms of compensatory beta-cell growth in insulin-resistant rats: roles of Akt kinase. Diabetes 54:2294–2304
Maeda H, Rajesh KG, Maeda H, Suzuki R, Sasaguri S (2004) Epidermal growth factor and insulin inhibit cell death in pancreatic beta cells by activation of PI3-kinase/AKT signaling pathway under oxidative stress. Transplant Proc 36:1163–1165
Zhao J, Zhang N, He M et al (2008) Increased beta-cell apoptosis and impaired insulin signaling pathway contributes to the onset of diabetes in OLETF rats. Cell Physiol Biochem 21:445–454
Peltier J, O'Neill A, Schaffer DV (2007) PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol 67:1348–1361
Ramnanan CJ, Edgerton DS, Kraft G, Cherrington AD (2011) Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes Metab 13(Suppl 1):S118–S125
Rivera N, Ramnanan CJ, An Z et al (2010) Insulin-induced hypoglycemia increases hepatic sensitivity to glucagon in dogs. J Clin Invest 120:4425–4435
Taniguchi CM, Tran TT, Kondo T et al (2006) Phosphoinositide 3-kinase regulatory subunit p85alpha suppresses insulin action via positive regulation of PTEN. Proc Natl Acad Sci U S A 103:12093–12097
Fisher SJ, Kahn CR (2003) Insulin signaling is required for insulin’s direct and indirect action on hepatic glucose production. J Clin Invest 111:463–468
Acknowledgements
The authors thank A. Klip (University of Toronto) for the pEtCAT vector and L6 muscle cell lines, X. Li, and N. Zhang (St Michael’s Hospital) for excellent technical support in performing RIAs, ELISA, and western blotting; A. Momen, J. Lee and T. Tong (University of Toronto) for conducting islet cell mass analysis.
Funding
This work was supported by grants from the Canadian Institutes for Health Research (CIHR) to Q. Wang (MOP-79534) and T. Jin (MOP-89987). Research in Q. Wang's laboratory was also supported by grants from the Canadian Diabetes Association (CDA) and the Juvenile Diabetes Research Foundation. Q. Wang was a CDA Scholar and is presently supported by the New Investigator Program from CIHR. L. Zhu and F. Huang were supported by the Chinese Overseas Scholarship Foundation.
Duality of interest statement
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
AM, LZ, TJ and QW designed, analysed and interpreted data and drafted the manuscript. AM, LZ, FH, HQ, HL, CYZ, TJ and QW analysed data and edited the manuscript. QW conceived and designed experiments, and edited the manuscript. All authors have approved the final version.
Author information
Authors and Affiliations
Corresponding author
Additional information
A. Maharaj and L. Zhu contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 83 kb)
ESM Fig. 2
(PDF 143 kb)
ESM Fig. 3
(PDF 14 kb)
ESM Fig. 4
(PDF 171 kb)
ESM Fig. 5
(PDF 58 kb)
ESM Fig. 6
(PDF 34 kb)
ESM Table 1
(PDF 84 kb)
Rights and permissions
About this article
Cite this article
Maharaj, A., Zhu, L., Huang, F. et al. Ectopic expression of glucagon receptor in skeletal muscles improves glucose homeostasis in a mouse model of diabetes. Diabetologia 55, 1458–1468 (2012). https://doi.org/10.1007/s00125-012-2464-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2464-x