
Abstract
Aims/hypothesis
Glycogen synthase kinase 3β (GSK-3β) is an enzyme that is suppressed by insulin and when elevated results in insulin resistance in skeletal muscle and diabetes. Its role in beta cell development and function is little known. Because of the enzyme’s anti-proliferative and pro-apoptotic properties, the hypothesis to be tested here was that beta cell specific deficiency of GSK-3β in mice would result in enhanced beta cell mass and function.
Methods
Mice with beta cell deficiency of GSK-3β (β-Gsk-3β [also known as Gsk3b]−/−) were generated by breeding Gsk-3β flox/flox mice with mice overexpressing the Cre recombinase gene under the control of the rat insulin 2 gene promoter (RIP-Cre mice), and glucose tolerance, insulin secretion, islet mass, proliferation and apoptosis were measured. Changes in islet proteins were investigated by western blotting.
Results
On a normal diet β-Gsk-3β –/– mice were found to have mild improvement of glucose tolerance and glucose-induced insulin secretion, and increased beta cell mass accompanied by increased proliferation and decreased apoptosis. On a high-fat diet β-Gsk-3β –/– mice exhibited improved glucose tolerance and expanded beta cell mass with increased proliferation relative to that in control mice, resisting fat-fed diabetes. Molecular mechanisms accounting for these phenotypic changes included increased levels of islet IRS1 and IRS2 proteins and phospho-Akt, suggesting enhanced signalling through the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, and increased islet levels of pancreas/duodenum homeobox protein 1 (PDX1). Inhibition of GSK3 in MIN6 cells in vitro led to increased IRS1 and IRS2 protein levels through inhibition of proteosomal degradation.
Conclusions/interpretation
These results are consistent with a mechanism whereby endogenous GSK-3β activity controls islet beta cell growth by feedback inhibition of the insulin receptor/PI3K/Akt signalling pathway.
Similar content being viewed by others

Introduction
The molecular mechanisms enabling beta cells to adapt to insulin resistance are being discovered primarily in animal models. A number of important genetic models have focused on the requirement for insulin signalling through beta cell insulin/IGF1 receptors, as reviewed in [1, 2]. In these models the primacy of phosphatidylinositol 3-kinase (PI3K)/Akt activity in expansion and postnatal maintenance of beta cell mass is apparent, as reviewed in [3]. The remarkable ability of beta cell mass to expand via enhanced proliferation and reduced apoptosis in transgenic mice producing constitutively active Akt in beta cells illustrates the potential importance of this pathway for expanding beta cells [4, 5]. Knowing that increased production of Akt in beta cells leads to marked expansion, our studies have focused on the role of an important negatively regulated Akt substrate, glycogen synthase kinase 3 (GSK3).
GSK3 was originally identified as a serine/threonine kinase that inactivates glycogen synthase [6]. This kinase is distinguished by the property of being constitutively active. Early studies showed that insulin inhibits GSK3 activity through PI3K/Akt-induced phosphorylation, promoting glycogen synthesis and glucose disposal. Later, many cellular processes, including transcription, translation, cell cycle regulation and apoptosis, were shown to be affected by GSK3 [6]. The adverse consequences of GSK3 activity have been illustrated by its overproduction in peripheral tissues in diabetic animals and patients [7–9], and by reversal of diabetes with GSK3 inhibitors [10]. While previous studies illustrated the undesirable consequences of overexpression of Gsk-3β (also known as Gsk3b) in peripheral tissues and islet beta cells, little is known about the consequences of reduced expression in islet beta cells. A recent in vitro study of GSK-3β activity in isolated human and rat islets showed that incubation with various chemical inhibitors resulted in enhanced DNA synthesis and cell cycle progression [11]. These in vitro studies suggested that this enzyme might be rate-limiting for islet beta cell growth in the animal in vivo. The hypothesis to be tested here was that deficiency of endogenous GSK-3β would result in expansion of beta cell mass through enhanced proliferation. The present experiments used a conditional Gsk-3β mouse (Gsk-3β flox/flox) to create mice with beta cell-specific GSK-3β deficiency (β-Gsk-3β –/–). The results of these studies now confirm that GSK-3β is rate-limiting for islet beta cell mass in vivo, and suggest a mechanism involving feedback inhibition of the insulin receptor/Akt-signalling pathway.
Methods
Animal production and phenotypic analysis
The generation of mice expressing a conditional allele of Gsk-3β was described previously [12]. Pancreatic beta cell-specific Gsk-3β knockout mice (β-Gsk3b –/–) were generated by breeding Gsk-3β floxed mice with mice that expressed the Cre recombinase gene under the control of the promoter of the rat insulin 2 gene (RIP-Cre mice) [13].The primers used for genotyping are listed in the Electronic supplementary material (ESM Table 1). For all studies, RIP-Cre mice were used as a control rather than wild-type mice because RIP-Cre mice have been shown to have glucose intolerance relative to wild-type mice [14]. The mice were fed a normal rodent diet (Picolab Rodent Diet 20, catalogue no. 5053; Purina Mills, St Louis, MO, USA) or a synthetic high-fat diet (HFD) (Teklad Adjusted Calories Diet; 55/fat, catalogue no. TD.93075; Harlan Laboratories, Indianapolis, IN, USA) and were housed in colony cages and maintained on a 12 h light–dark cycle. The composition of the standard diet was 20% protein, 4.5% fat. The composition of the HFD diet was 21.2% protein, 54.8% fat from vegetable shortening.
Blood glucose and serum insulin concentrations were determined as previously described [4]. This project was approved by the Animal Ethics Committee of Washington University School of Medicine.
Immunohistochemical and morphometric analysis of pancreatic islets
Pancreases were isolated from 3-month-old RIP-Cre control and β-Gsk-3β −/− mice and fixed. Immunohistological and morphometric analyses were as described previously [15]. Antibodies used in this study are listed in the Electronic supplementary material (ESM) Table 1. The mass of beta cells was quantified using NIH Image 1.39j software [16]. Islet beta cell proliferation and apoptosis were assessed as described previously [15]. Quantitative data were obtained from at least three mice in each group unless indicated otherwise.
Assays of islet insulin secretion, insulin content and DNA content
Glucose-stimulated insulin secretion and insulin content were determined in islets isolated by collagenase distension/disgestion of the pancreas, as described previously [17].
Western blotting analysis and cell culture
Islets were isolated as described previously [18] and immunoblots were analysed as described previously [15]. Extracts (20 μg total protein) were subjected to immunoblot analysis with antibodies (for specific antibodies see ESM Table 1). Immune complexes were revealed using the ECL Advance Western Blot Detection Kit (Amersham, Little Chalfont, UK) and images were acquired using a FluoroChemi 8800 digital camera acquisition system (Alpha Innotech, San Leandro, CA, USA). Band intensities in the blots were later quantified using ImageJ 1.39j [16] and α-tubulin or β-actin bands were used to adjust for loading differences.
MIN6 cells were maintained in DMEM [19]. Cells with passage numbers <35 were used. For immunoblot analysis, we prepared lysates of MIN6 cells treated with either 20 mmol/l LiCl or 20 mmol/l NaCl for 30 min, then cotreated them with 50 μg/ml cycloheximide and 5 μmol/l MG132 (Calbiochem, San Diego, CA, USA) or cotreated them with DMSO and either 0.5% BSA or 400 μmol/l palmitate+0.5% BSA at the indicated time points. The lysates were probed with the antibodies listed above. Protein was extracted with a cell lysis buffer [15]. Protein samples (20 μg) were subjected to immunoblot analysis with antibodies to IRS1 and IRS2 (Upstate, Lake Placid, NY, USA).
Immunoprecipitation
MIN6 cell lysates (1 mg) were incubated with antibody specific for IRS2 or IRS1 (Upstate, Lake Placid, NY, USA) overnight at 4°C with gentle agitation. Protein A/G beads were then added and incubation was continued for 4 h at 4°C. Immunoprecipitated proteins were subjected to SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with anti-phosphotyrosine antibody (Upstate, Lake Placid, NY, USA).
Statistical analysis
Quantitative data are presented as mean ± SEM from at least three independent experiments and at least 100 islets from more than three mice unless indicated otherwise. We assessed interactions among variables by two-way analysis of variance and used the Student’s t test to compare independent means, or the Wilcoxon–Mann–Whitney U test to compare means when sample sizes differed. A p value of <0.05 was considered statistically significant.
Results
Generation of beta cell-specific Gsk-3β knockout mice
The derivation of Gsk3b flox/flox mice has been described previously [12]. We first generated mice with beta cell deficiency of GSK-3β (β-Gsk-3β –/–) by breeding Gsk-3β flox/flox mice with mice overexpressing the Cre recombinase gene under the control of the rat insulin 2 gene promoter (RIP-Cre mice) [13]. Immunoblot analysis of islet protein lysates with an antibody that recognises both forms of GSK3 (GSK-3α and GSK-3β) indicated that GSK-3β appeared to be the major form (Fig. 1a). In β-Gsk-3β –/– mice the amount of GSK-3β was reduced by more than 80% compared with control mice (p < 0.05). There appeared to be no compensatory increase in GSK-3α protein levels. Phosphorylation of glycogen synthase, a substrate of GSK-3β and a measure of GSK-3β activity, was estimated to be less than 50% of that in control animals. This probably represents a minimal estimate of the loss of GSK-3β activity in beta cells since this was analysed on total islet protein including cells other than beta cells. Immunochemical analyses of pancreatic sections from 12-week-old mice indicated that GSK-3β was almost non-existent in beta cells of β-Gsk-3β –/– compared with RIP-Cre control mice (Fig. 1b). Double antibody staining for GSK-3β and glucagon indicated that GSK-3β was present predominantly in beta cells (ESM Fig. 1).

Generation of beta cell-specific Gsk-3β knockout (β-Gsk-3β –/–) mice. a Islets were isolated from RIP-Cre control and β-Gsk-3β –/– mice and protein lysates were subjected to western blot analysis using anti-total GSK-3α/β, anti-phospho-glycogen synthase (Phospho GS) and anti-α-tubulin. One of three independent experiments is shown. b The GSK-3β level was quantified by densitometry and normalised to α-tubulin. *p < 0.05. c Immunohistochemical analysis of GSK-3β on representative pancreatic sections from β-Gsk-3β –/– mice and RIP-Cre control mice. Anti-GSK-3β antibody was labelled with cys (red) and anti-insulin antibody with FITC (green), indicating the markedly reduced abundance of GSK-3β in β-Gsk-3β –/– mice
Tissue-specific deletion of GSK-3β was demonstrated by observing that the abundance of GSK-3β was similar in the brain, hypothalamus, adipose tissue, liver, skeletal muscle and heart in β-Gsk-3β –/– and control mice (ESM Fig. 2), consistent with previous observations [20].
Metabolic characteristics of β-Gsk-3β –/– mice
We next examined whether reduction of GSK-3β in beta cells resulted in metabolic alterations. There were no significant differences in body weight during the 26 weeks of observation (ESM Fig. 3a) or in food intake (ESM Fig. 3b). The fasting and fed glucose levels were lower in β-Gsk-3β –/– mice compared with RIP-Cre control mice, while insulin levels were not different (Table 1, ESM Fig. 4). Intraperitoneal glucose tolerance testing of β-Gsk-3β –/– and control mice at 12 and 24 weeks of age revealed that β-Gsk-3β –/– mice had lower fasting blood glucose and improved glucose tolerance at both age points. The fasting blood glucose concentration at 12 weeks of age was 3.73 ± 0.22 mmol/l in β-Gsk-3β –/– mice and 5.19 ± 0.15 mmol/l in RIP-Cre control mice (p < 0.05) (Fig. 2a). The insulin response 3 and 10 min after intraperitoneal glucose injection was also examined in mice at 12 weeks of age. The plasma insulin level was significantly higher in β-Gsk-3β –/– mice than in control mice at 10 min (Fig. 2b).

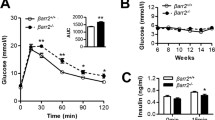
Glucose metabolism and glucose-induced insulin secretion in β-Gsk-3β –/– mice. Glucose tolerance tests were performed on overnight-fasted male RIP-Cre control and β-Gsk-3β –/– mice at 12 weeks (a) and 24 weeks (b) of age after intraperitoneal injection of d-glucose (2 g/kg body weight; n = 6). Glucose levels were assessed at the indicated time intervals. Results are presented as mean ± SEM; *p < 0.05, **p < 0.01. c Plasma insulin levels were measured before (0 min) and at the indicated time points after intraperitoneal injection of glucose (2 g/kg body weight) following an overnight fast (n = 5). Grey lines, RIP-Cre control mice; black lines, β-Gsk-3β –/– mice
Morphology of islets of β-Gsk-3β –/– mice
To assess a possible mechanism for the increased insulin secretion, the effect of GSK-3β deficiency on islet morphology was next examined. Beta cell mass in β-Gsk-3β –/– mice at 12 weeks of age was approximately 25% higher than in control mice, although the representative image shown appears to show a larger difference (p < 0.05; Fig. 3a). The proliferation rate was addressed by double immunostaining for Ki67 and insulin (Fig. 3b). Relative Ki67 production in the beta cells of the β-Gsk-3β –/– mice was increased more than twofold compared with controls (p < 0.05; Fig. 3b). Apoptosis was then assessed by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labelling (TUNEL) staining in pancreatic beta cells at 12 weeks of age (Fig. 3c). The number of TUNEL-positive beta cells was reduced by approximately 60% in β-Gsk-3β –/– mice compared with control mice. The combination of improved proliferation and reduced apoptosis can account for the higher beta cell mass in βGsk3b –/– mice, and correlates with the increased glucose tolerance and insulin secretion.

Islet morphology in β-Gsk-3β –/– mice. a Beta cell mass in RIP-Cre control and β-Gsk-3β –/– mice. Pancreatic sections from 12-week-old mice of the indicated genotypes were stained with antibodies to insulin and then counterstained with haematoxylin. Scale bar, 100 μm. Random sections of the entire pancreas were stained with haematoxylin and were measured to determine beta cell mass (n = 4 for each genotype). Results are expressed as beta cell mass (mg), shown as mean ± SEM (b). *p < 0.05. c Pancreatic sections from 12-week-old mice of the indicated genotypes were stained with antibodies to Ki67 (red) and insulin (green). Arrowheads indicate proliferating cells. Scale bar, 50 μm. d The numbers of cells that are positive for both Ki67 and insulin were quantified as percentages of the total number of insulin-positive cells in the sections (n = 4 for each genotype). Results are mean ± SEM. *p < 0.05. e Apoptotic beta cells in pancreatic sections from 12-week-old mice of the indicated genotypes (n = 3 for each genotype) were assessed by TUNEL and anti-insulin staining. The proportion of cells that were positive for both is shown as the percentage of the total number of insulin-positive cells in the section. Results are mean ± SEM. *p < 0.05. f In vitro analysis of glucose-stimulated insulin secretion according to DNA content of isolated islets. Similar sized islets (n = 5) from mice at 12 weeks of age (n = 4 for each genotype) were sequentially incubated in 1 ml Krebs-Ringer bicarbonate HEPES buffer containing 2.8 mmol/l glucose or 16.8 mmol/l glucose for 60 min at 37°C, with preincubation at each interval in 2.8 mmol/l glucose buffer for 30 min. Insulin released was determined by radioimmunoassay and corrected for islet DNA content. † p = 0.06. g Insulin content in isolated islets of RIP-Cre control and β-Gsk-3β –/– mice at 12 weeks of age. Insulin was extracted by the acid–ethanol method from four mice of each genotype and was measured by radioimmunoassay
To assess another possible mechanism for the improved glucose tolerance and glucose-stimulated insulin secretion, the effects of islet GSK-3β deficiency on insulin secretion from isolated islets was examined. As shown in Fig. 3d, insulin release from islets of β-Gsk-3β –/– mice at 16.8 mmol/l glucose appeared to be increased compared with that of islets of control mice, although the increase was not significant (p = 0.06). Islet insulin contents of β-Gsk-3β –/– mice were indistinguishable from those of control mice (Fig. 3e). Thus, the increased insulin levels in β-Gsk-3β –/– mice may be due to increased insulin secretion per islet as well as increased islet mass.
Signalling mechanisms underlying expanded beta cell mass and function in β-Gsk-3β –/– mice
Numerous studies have documented the role of insulin signalling through the insulin receptor/PI3K/Akt pathway in the expansion of beta cell mass, as reviewed in [1, 2, 21]. Tissue-specific reduction of skeletal muscle GSK-3β in mice resulted in enhanced insulin signalling and glucose disposal in vivo [12]. Analysis of skeletal muscle cells in vitro showed that treatment with a chemical inhibitor of GSK3 resulted in an increase in insulin-stimulated glucose uptake associated with increased IRS1 protein [22]. To determine whether pancreatic beta cell GSK-3β deficiency was altering the insulin receptor/Akt signalling pathway through suppression of IRS protein levels in control compared with β-Gsk-3β –/– mice, the key proteins of this signalling pathway were examined in isolated islets from mice at 12 weeks of age. IRS protein levels and phosphorylation of Akt/PKB were evaluated as downstream measures. As shown in Fig. 4a, there was an increase of >70% in IRS2 protein level in islets of β-Gsk-3β –/– mice (p < 0.05). IRS1 protein levels in islets were also increased by >70% in β-Gsk-3β –/– mice compared with those of control mice (p < 0.01). Consistent with an increase in IRS protein levels and suggestive of enhanced signalling activity, Akt activity appeared to be enhanced, with a 70% increase in phosphorylation of serine 473 (p < 0.05). Phospho-forkhead box O1 (FOXO1) appeared to be increased in β-Gsk-3β –/– mice relative to RIP-Cre mice, consistent with the increased p-Akt, although this was not quantified. A comparable increase (p < 0.05) in pancreas/duodenum homeobox protein 1 (PDX1) protein in islets of β-Gsk-3β –/– mice provided further support for enhanced insulin signalling [23].

Proteins mediating insulin receptor/Akt signalling in pancreatic islets of β-Gsk-3β –/– mice. a Islets were isolated from 12-week-old male RIP-Cre control and β-Gsk-3β –/– mice and lysates were subjected to western blot analysis using antibodies to the indicated proteins. One of five independent experiments is shown. Levels of IRS1 and IRS2 and PDX1 were quantified by densitometry and normalised to α-tubulin. Phosphorylation of serine 473 of Akt was normalised by total Akt. Protein and phosphorylation levels are shown as mean ± SEM. b IRS2, c IRS1, d PDX1, e pAkt(S473); *p < 0.05, **p < 0.01. f Islets were isolated from 12-week-old male wild-type mice and were serum-deprived for 6 h and then treated with either the GSK3 inhibitor SB216763 or DMSO as a vehicle for 16 h in RPMI medium. Total cell lysates were subjected to western blot analysis using antibodies to the indicated proteins. One of three independent experiments is shown. Densitometric data on IRS1 and IRS2 and PDX1 were normalised to α-tubulin. Phosphorylation of serine 473 of Akt was normalised for total Akt. Protein and phosphorylation levels are shown as mean ± SEM. g IRS2, h IRS1, i PDX1, j pAkt(S473); *p < 0.05
Further support for the hypothesis that reduced GSK-3β activity in β-Gsk-3β –/– mice can increase IRS1 and IRS2 levels and insulin signalling was obtained by in vitro experiments with a potent and selective inhibitor of GSK-3 activity, SB216763 [24]. Islets were isolated from 12-week-old male wild-type mice, serum-deprived for 6 h, then treated with either SB216763 or DMSO as a vehicle for 16 h in RPMI medium. Western blot analysis of cell lysates showed that inhibition of GSK3 was associated with enhanced levels of IRS1 and IRS2 and PDX1 protein, along with increased phospho-Akt (Fig. 4b), findings consistent with those obtained in islets isolated from β-Gsk-3β –/– mice.
GSK-3β has been shown to phosphorylate several intracellular components of the Wnt signal transduction pathway [25]. It is well known that wingless-type MMTV integration site family member 2 (WNT) signalling regulates cellular proliferation and development. GSK-3β activity promotes phophorylation of β-catenin and protein degradation. As shown in ESM Fig. 5a, active β-catenin measured with antibody specific for dephosphorylation at serine 37 and threonine 41 [26] was increased in islets from β-Gsk-3β –/– mice relative to control mice. These results are consistent with activation of β-catenin as a contributor to the observed expanded beta cell mass in β-Gsk-3β –/– mice.
The effects of GSK-3β activity on cell cycle regulatory proteins in beta cells have been little studied. Irs2 knockout mice have been shown to have fourfold elevation of GSK-3 activity in islets that is associated with an elevated level of the cell cycle inhibitor p27Kip1 and GSK-3 activity in insulinoma cells has been shown to stabilise p27 protein [15]. Similarly, in other cell types GSK-3 activity has been shown to regulate degradation, for example the stability of p27 [15, 27]. The protein levels of the cyclin-dependent kinase inhibitors p27kip1 and p21cip1 in islets from β-Gsk-3β –/– mice did not appear to be altered (ESM Fig. 5b).
Evidence that GSK3B inhibition increases insulin signalling and reduces proteasomal degradation of IRS proteins in vitro
Reduced GSK-3β activity in β-Gsk-3β –/– mice resulted in increases in islet IRS1 and IRS2 proteins, phospho-Akt, PDX1 protein and beta cell mass, but the molecular mechanisms are unknown. A potential mechanism would be that GSK-3β has a direct effect on insulin signalling through phosphorylation of the IRS proteins. Because the putative phosphorylation sites of IRS1 and IRS2 by GSK-3 are unknown, we devised an in vitro experiment using the strategy of Dokken et al. [28] in which the effect of GSK-3β inhibition on insulin signalling in skeletal muscle was demonstrated. MIN6 cells were incubated with the GSK-3 inhibitor SB216763 to inhibit the activity of GSK-3β. Following immunoprecipitation of IRS1 and IRS2 proteins, immunoblotting with antibody to phosphotyrosine demonstrated a significant increase in phosphorylation of both proteins (Fig. 5a). These results are consistent with the inhibitory effect of GSK-3β on insulin signalling.

GSK-3β activity affects protein stability of IRS1 and IRS2 in MIN6 cells. a, b MIN6 cells were cultured for 6 h in medium with 5 mmol/l glucose without serum and then treated with either the GSK3 inhibitor SB216763 or DMSO as a vehicle for 16 h. Total cell lysates were immunoprecipitated (IP) with IRS2 (a) or IRS1 (b) antibody. Precipitates were subjected to western blotting with anti-phosphotyrosine [P(Y)] antibody. Phosphorylation levels are shown as mean ± SEM. *p < 0.05. c MIN6 cells were cultured overnight in medium with 5 mmol/l glucose without serum. The following day, the cells were pretreated with either 20 mmol/l LiCl or 20 mmol/l NaCl as control for 30 min before addition of 50 μg/ml cycloheximide to all samples. Where indicated, either MG132 (5 μmol/l) or DMSO as vehicle control for MG132 was added for 4 h. Also where indicated, fatty acid (FA; 400 μmol/l palmitate+0.5% BSA) or 0.5% BSA was added for 4 h. Total cell lysates were obtained at the indicated time points and were subjected to western blotting with IRS1, IRS2 and α-tubulin antibodies. One of three independent experiments is shown. Protein levels at indicated time points were normalised to α-tubulin and are shown as mean ± SEM (d IRS2, e IRS1). *p < 0.05. White columns, control; light grey columns, LiCl; black columns, fatty acid; dark grey columns, LiCl+fatty acid
After phosphorylation by GSK-3, a number of proteins have been demonstrated to be regulated at the post-translational level by either enhancing stability [27] or increasing degradation [29, 30]. One of the mechanisms whereby GSK-3 activity increases insulin resistance in skeletal muscle is by phosphorylating and promoting proteasomal degradation of IRS1 [28], and in liver by a similar post-translational reduction in IRS2 [31]. An increased IRS protein level in islets of β-Gsk-3β –/– mice could be accounted for by effects of GSK-3β activity on the stability of proteins. An experiment was designed to test this hypothesis.
Mouse insulinoma cells (MIN6) were treated with cycloheximide to block new protein synthesis and with either LiCl to inhibit GSK-3β activity or MG132 to inhibit proteasome-mediated protein degradation [29]. Fatty acid treatment of MIN6 insulinoma cells was shown to induce endoplasmic reticulum stress and reduce insulin receptor/Akt signalling [32], so cotreatment with fatty acid (palmitate 400 μmol/l) for 4 h was used in an attempt to induce endoplasmic reticulum stress and further activate GSK-3β activity. Cell lysates were analysed by western blotting, as shown in Fig. 5b. Addition of cycloheximide for 4 h resulted in a reduction of approximately 30–35% in IRS1 and IRS2 protein levels. The addition of LiCl had a significant effect on the preservation of these proteins. Addition of fatty acid resulted in further reduction of IRS1 and IRS2 levels, and again LiCl addition significantly increased protein levels. MG132 cotreatment preserved both the IRS1 and the IRS2 protein level. These results suggest that endogenous GSK-3β regulates IRS1 and IRS2 protein levels in beta cells through proteasome-mediated degradation.
β-Gsk-3β –/– mice on a high-fat diet exhibited resistance to diabetes
Obesity is a major cause of diabetes in humans because of insulin resistance and subsequent beta cell dysfunction [33]. In experimental animals, an HFD results in insulin resistance, hyperinsulinaemia and expanded beta cell mass [34]. However, beta cell islet hyperplasia is not sufficient to prevent glucose intolerance. On a normal diet, β-Gsk-3β –/– mice developed expanded beta cell mass associated with increased proliferation and decreased apoptosis. To determine whether endogenous GSK-3β is rate-limiting for expansion of beta cell mass under conditions of HFD and whether β-Gsk-3β –/– mice would resist fat feeding-induced diabetes, β-Gsk-3β –/– and RIP-Cre mice were fed a diet containing 55% fat or a normal diet for up to 12 weeks. The β-Gsk-3β –/– mice and control mice gained body weight similarly on both diets (data not shown). After 12 weeks of high-fat feeding, glucose intolerance was noted in RIP-Cre control animals, while in contrast it was significantly improved in β-Gsk-3β –/– mice (Fig. 6a). In the β-Gsk-3β –/– mice, along with resistance to fat feeding-induced diabetes, insulin secretion following glucose challenge was enhanced relative to that in control animals (Fig. 6b). Consistent with the improved metabolic phenotype of β-Gsk-3β –/– mice on the HFD, we observed that β-Gsk-3β –/– mice had expanded beta cell mass relative to control mice after the HFD (Fig. 6c). This increased beta cell mass was associated with increased beta cell proliferation in β-Gsk-3β –/– mice relative to control mice on the HFD (Fig. 6d). Apoptosis was then assessed by TUNEL staining in pancreatic beta cells at 24 weeks of age after 12 weeks on the HFD (Fig. 6e). The number of TUNEL-positive beta cells was reduced by approximately 70% in β-Gsk-3β –/– mice compared with control mice. The combination of improved proliferation and reduced apoptosis can account for the higher beta cell mass in β-Gsk-3β –/– mice, and correlated with the enhanced glucose tolerance and insulin secretion. These results indicate that β-Gsk-3β –/– mice resist fat feeding-induced diabetes and that endogenous GSK-3β is rate-limiting for the expansion of beta cell mass under these conditions.

Effects of a high-fat diet (HFD) on β-Gsk-3β –/– mice. RIP-Cre control and β-Gsk-3β –/– mice were fed a HFD diet for 12 weeks. a After fat feeding, intraperitoneal glucose tolerance tests were performed on overnight-fasted male RIP-Cre control (n = 5) and β-Gsk-3β –/– mice (n = 5). Glucose levels were assessed at the indicated time intervals. Results are mean ± SEM. *p < 0.05. Grey line, RIP-Cre control; black line, β-Gsk-3β –/–. b Plasma insulin levels were measured before (0 min) and at the indicated time points after intraperitoneal injection of glucose (2 g/kg of body weight) following an overnight fast (RIP-Cre control, n = 4; β-Gsk-3β –/– n = 5). Grey line, RIP-Cre control; black line, β-Gsk-3β –/–. c Beta cell mass in RIP-Cre control and β-Gsk-3β –/– mice after 12 weeks of high-fat feeding. Pancreatic sections from mice of the indicated genotypes that were fed a high-fat diet for 12 weeks were stained with antibodies to insulin and then counterstained with haematoxylin. Scale bar, 100 μm. Random sections of the entire pancreas from mice of the indicated genotypes were stained with haematoxylin and were measured to determine beta cell mass (RIP-Cre control, n = 4; β-Gsk-3β –/– , n = 4). Results are expressed as beta cell mass (mg). All values are mean ± SEM. **p < 0.01. d Measurement of Ki67-positive beta cells. Arrowheads indicate proliferating cells. The numbers of cells positive for both Ki67 and insulin are quantified as the percentage of the total number of insulin-positive cells in the sections. (RIP-Cre control, n = 4; β-Gsk-3β –/–, n = 4). Results are mean ± SEM. *p < 0.05. Scale bar, 50 μm. e Apoptotic beta cells in pancreatic sections from 24-week-old mice of the indicated genotypes that were fed an HFD diet for 12 weeks (n = 3 for each genotype) were assessed by TUNEL and anti-insulin staining. Proportions of cells that were positive for both are represented as the percentages of the total number of insulin-positive cells in the sections. Results are mean ± SEM. *p < 0.05
Discussion
Previous studies of islets with chemical inhibitors of GSK-3β suggested that this enzyme might be rate-limiting for beta cell proliferation [11]. In the present study we sought to examine the hypothesis that GSK-3β is rate-limiting for islet beta cell mass and function, and that targeted deletion in β-Gsk-3β –/– mice would result in expanded beta cell mass and function. These mice were found to have improved glucose tolerance, enhanced glucose-induced insulin secretion, and increased beta cell mass accompanied by enhanced proliferation and decreased apoptosis. Additionally, β-Gsk-3β –/– mice on a high-fat diet exhibited resistance to fat feeding-induced glucose intolerance relative to control mice. Molecular mechanisms accounting for these phenotypic changes in chow-fed mice included increased levels of islet IRS1 and IRS2 proteins and phospho-Akt, suggesting enhanced signalling through the insulin receptor/PI3K/Akt pathway and increased islet PDX1 levels. Inhibition of GSK3 in MIN6 cells in vitro led to increased IRS1 and IRS2 protein through inhibition of proteasomal degradation. These results are consistent with a mechanism whereby endogenous GSK-3β activity controls islet beta cell growth by feedback inhibition of the insulin receptor/PI3K/Akt signalling pathway.
An increase of more than 70% in islet IRS2 protein levels in β-Gsk-3β –/– mice relative to control mice on a normal diet was noted (Fig. 4a). The importance of IRS2 protein to islet beta cell function was established with IRS2-deficient mice that developed profound diabetes with severe loss of beta cells at a young age [35]. Our finding of increased IRS2 and phospho-Akt in β-Gsk-3β –/– mice (Fig. 4a), along with in vitro evidence in MIN6 cells that GSK-3β phosphorylates IRS2 and targets it for proteasome-mediated degradation (Fig. 5b), suggests a possible inhibitory mechanism for GSK-3β in insulin receptor/PI3K/Akt signalling in islet beta cells. An increase of more than 70% in islet IRS1 protein levels in β-Gsk-3β –/– mice relative to control mice was also noted, although the role of IRS1 in islet beta cell function relative to that of IRS2 is less clear.
The expanded beta cell mass in β-Gsk-3β –/– mice was accompanied by enhanced proliferation and decreased apoptosis, and one of the effectors could be PDX1. Previous in vitro experiments in insulinoma cells showed that GSK-3 activity phosphorylates PDX1 and promotes its proteasomal degradation [29]. In the present in vivo study we observed in βGsk-3β –/– mice a significant increase in islet PDX1 protein level relative to that in control mice, consistent with the reduction in PDX1 degradation and with increased insulin receptor/PI3K/Akt signalling. We did not measure PDX1 phosphorylation, however, as an antibody to the serine site phosphorylated by GSK3 was not available. Another in vivo example of GSK-3β regulation of PDX1 levels was observed in Irs2 −/− mice rescued by crossing with Gsk-3β-haploinsufficient mice [15]. Other proteins known to regulate islet beta cell proliferation include β-catenin and D-cyclins, and GSK-3 activity is well known to affect the levels of these proteins [11]. In the present study preliminary experiments showed that islets of β-Gsk-3β –/– mice had an increase in active β-catenin (ESM Fig. 5a), which may have contributed to the expansion of beta cell mass.
The phenotypes observed in β-Gsk-3β –/– mice leave unanswered the question of the role of islet GSK3A in beta cell development and function. Conditional deletion of the Gsk-3α (also known as Gsk3a) gene in islet beta cells may result in further expansion of mass and function, but this has yet to be determined.
Numerous studies have reported insulin resistance in animals fed an HFD. We compared RIP-Cre control with β-Gsk-3β –/– mice on both a normal diet and an HFD. Fasting plasma glucose levels in β-Gsk-3β –/– and RIP-Cre mice on the normal diet and the HFD are shown for comparison in ESM Fig. 4. Results of an intraperitoneal glucose tolerance test (IPGTT) showed that on the HFD the glucose tolerance of β-Gsk-3β –/– mice at 24 weeks of age was significantly improved relative to RIP-Cre control mice at the same age (Fig. 6a) on the HFD. We also used the area under the curve to assess IPGTT results (ESM Fig. 6a, b). The results showed that, on the normal diet, blood glucose was about 31% lower in β-Gsk-3β –/– mice compared with RIP-Cre control mice, and on the HFD it was reduced by about 27% in β-Gsk-3β –/– mice compared with RIP-Cre controls (ESM Fig. 6a, b). We also compared the beta cell mass of β-Gsk-3β –/– and RIP-Cre mice on the two diets, although the data for the normal diet were on 12-week-old mice while those for the HFD were on 24-week-old mice. The beta cell mass of βGsk3b –/– mice on the normal diet was 22% higher than that of RIP-Cre control mice on the normal diet, while on the HFD the beta cell mass of β-Gsk-3β –/– mice was 45% higher than that of RIP-Cre control mice. These results indicate that β-Gsk-3β –/– mice resist fat feeding-induced diabetes at least in part as a result of increased beta cell mass and proliferation.
A number of genetic models that alter the insulin signalling pathway in mice have provided substantial evidence that this pathway is a major contributor to beta cell mass and islet function. Signalling through the PI3K/Akt pathway has been shown to be critical for the eventual outcome of beta cell mass and function. There are at least three major targets of this signalling, including mechanistic target of rapamycin (serine/threonine kinase) (mTOR)/S6 kinase, FOXO and GSK-3. The present study provides evidence that GSK-3β is rate-limiting for beta cell growth by feedback inhibition of the insulin signalling pathway through its inhibitory effects on IRS1 and IRS2. There are few direct data on FOXO activity in pancreatic islet beta cells. In vivo studies with Irs2 −/− mice showed reduced beta cell mass and diabetes, and crossing these mice with mice haploinsufficient for Foxo1 restored beta cell mass and proliferation in these mice [36]. An acute in vitro study using an adenovirus expressing a gain of function form of FOXO showed decreased beta cell glucose utilisation and insulin secretion [37].
While both GSK-3β and FOXO appear to have negative effects on beta cell proliferation and mass, mTOR complex 1 (mTORC1)/S6 kinase 1 signalling appears to have the opposite effect. In vitro studies with rapamycin showed inhibition of proliferation [11], and in vivo knockout of S6k1 (also known as S6k) resulted in reduced beta cell mass and diabetes [38]. Thus, the present study shows that GSK-3β is only one part of the insulin-signalling pathway controlling islet beta cell mass and function.
In skeletal muscle and liver GSK-3β is constitutively active, and in the fasted state with low insulin levels this enzyme inhibits glycogen storage. With feeding, insulin secretion inhibits the enzyme, facilitating glycogen synthesis. In the present study, tissue-specific ablation of GSK-3β in pancreatic islet beta cells resulted in a phenotype that suggested the normal function of this enzyme. In contrast to peripheral tissues, the present findings suggest that GSK-3β controls the expansion of beta cell proliferation in the islet. The finding that absence of the enzyme in beta cells during the stress of fat feeding results in normal glucose tolerance and enhanced beta cell mass indicates that this enzyme is a potentially important therapeutic target for treating diet-induced diabetes in man.
Abbreviations
- FOXO:
-
Forkhead box O
- GSK-3β:
-
Glycogen synthase kinase 3β
- HFD:
-
High-fat diet
- IPGTT:
-
Intraperitoneal glucose tolerance test
- PDX1:
-
Pancreas/duodenum homeobox protein 1
- PI3K:
-
Phosphatidylinositol 3-kinase
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labelling
References
Neubauer N, Kulkarni RN (2006) Molecular approaches to study control of glucose homeostasis. ILAR J 47:199–211
Kulkarni RN (2005) New insights into the roles of insulin/IGF-I in the development and maintenance of beta-cell mass. Rev Endocr Metab Dis 6:199–210
Heit JJ, Karnik SK, Kim SK (2006) Intrinsic regulators of pancreatic beta-cell proliferation. Annu Rev Cell Dev Biol 22:311–338
Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA (2001) Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest 108:1631–1638
Tuttle RL, Gill NS, Pugh W et al (2001) Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med 7:1133–1137
Kockeritz L, Doble B, Patel S, Woodgett JR (2006) Glycogen synthase kinase-3—an overview of an over-achieving protein kinase. Curr Drug Targets 7:1377–1388
Eldar-Finkelman H, Krebs EG (1997) Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci USA 94:9660–9664
Eldar-Finkelman H, Schreyer SA, Shinohara MM, LeBoeuf RC, Krebs EG (1999) Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes 48:1662–1666
Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR (2000) Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes 49:263–271
Rao R, Hao CM, Redha R, Wasserman DH, McGuinness OP, Breyer MD (2007) Glycogen synthase kinase 3 inhibition improves insulin-stimulated glucose metabolism but not hypertension in high-fat-fed C57BL/6J mice. Diabetologia 50:452–460
Liu H, Remedi MS, Pappan KL et al (2009) Glycogen synthase kinase-3 and mammalian target of rapamycin pathways contribute to DNA synthesis, cell cycle progression, and proliferation in human islets. Diabetes 58:663–672
Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR (2008) Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol 28:6314–6328
Herrera PL (2000) Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 127:2317–2322
Lee JY, Ristow M, Lin X, White MF, Magnuson MA, Hennighausen L (2006) RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J Biol Chem 281:2649–2653
Tanabe K, Liu Z, Patel S et al (2008) Genetic deficiency of glycogen synthase kinase-3-beta corrects diabetes in mouse models of insulin resistance. PLoS Biol 6:e37
Girish V, Vijayalakshmi A (2004) Affordable image analysis using NIH Image/ImageJ. Indian J Cancer 41:47
Martinez SC, Cras-Meneur C, Bernal-Mizrachi E, Permutt MA (2006) Glucose regulates Foxo1 through insulin receptor signaling in the pancreatic islet β-cell. Diabetes 55:1581–1591
Bernal-Mizrachi E, Fatrai S, Johnson JD et al (2004) Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest 114:928–936
Ohsugi M, Cras-Meneur C, Zhou Y et al (2005) Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. J Biol Chem 280:4992–5003
Heit JJ, Apelqvist AA, Gu X et al (2006) Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 443:345–349
White MF (2006) Regulating insulin signaling and beta-cell function through IRS proteins. Can J Physiol Pharmacol 84:725–737
Nikoulina SE, Ciaraldi TP, Mudaliar S, Carter L, Johnson K, Henry RR (2002) Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes 51:2190–2198
Kushner JA, Ye J, Schubert M et al (2002) Pdx1 restores beta cell function in Irs2 knockout mice. J Clin Invest 109:1193–1201
Coghlan MP, Culbert AA, Cross DA et al (2000) Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol 7:793–803
Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A (1998) Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J 17:1371–1384
Amit S, Hatzubai A, Birman Y et al (2002) Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev 16:1066–1076
Surjit M, Lal SK (2007) Glycogen synthase kinase-3 phosphorylates and regulates the stability of p27kip1 protein. Cell Cycle 6:580–588
Dokken BB, Sloniger JA, Henriksen EJ (2005) Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in prediabetic insulin-resistant rat skeletal muscle. Am J Physiol Endocrinol Metab 288:E1188–E1194
Boucher MJ, Selander L, Carlsson L, Edlund H (2006) Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J Biol Chem 281:6395–6403
Guo S, Burnette R, Zhao L et al (2009) The stability and transactivation potential of the mammalian MafA transcription factor are regulated by serine 65 phosphorylation. J Biol Chem 284:759–765
Sharfi H, Eldar-Finkelman H (2008) Sequential phosphorylation of insulin receptor substrate-2 by glycogen synthase kinase-3 and c-Jun NH2-terminal kinase plays a role in hepatic insulin signaling. Am J Physiol Endocrinol Metab 294:E307–E315
Martinez SC, Tanabe K, Cras-Meneur C, Abumrad NA, Bernal-Mizrachi E, Permutt MA (2008) Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes 57:846–859
Horton ES (2009) Effects of lifestyle changes to reduce risks of diabetes and associated cardiovascular risks: results from large scale efficacy trials. Obesity (Silver Spring) 17(Suppl 3):S43–S48
Terauchi Y, Takamoto I, Kubota N et al (2007) Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest 117:246–257
Withers DJ, Gutierrez JS, Towery H et al (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391:900–904
Kitamura T, Nakae J, Kitamura Y et al (2002) The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest 110:1839–1847
Buteau J, Shlien A, Foisy S, Accili D (2007) Metabolic diapause in pancreatic beta-cells expressing a gain-of-function mutant of the forkhead protein Foxo1. J Biol Chem 282:287–293
Um SH, Frigerio F, Watanabe M et al (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431:200–205
Acknowledgements
The authors would like to thank other members of Dr Permutt’s lab for helpful discussions. The Washington University Diabetes Research Training Center (P60DK020579-30) is acknowledged for assistance from the Immunoassay and Morphology Cores and the Adipocyte Biology Core, Clinical Nutrition Research Unit (NIH DK56351).
Funding
This work was supported by National Institutes of Health grants (R37 DK16746, to MAP) and the Canadian Institutes of Health Research (J. Woodgett) and Grants-in-Aid for Scientific Research (grant no. 22590984, to K. Tanabe) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. K. Tanabe was funded by the Takeda Science Foundation. Y. Liu was supported by an American Diabetes Association Mentor Based Fellowship Award.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Y. Liu and K. Tanabe contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Immunohistochemical analysis of GSK-3β and glucagon on representative pancreatic sections from β-Gsk-3β –/– mice and RIP-Cre control mice. Anti-GSK3B antibody was labelled with cys (red) and anti-glucagon antibody with FITC (green), indicating the markedly reduced abundance of GSK-3β in beta cells and that GSK-3β was not affected in alpha cells in β-Gsk3b –/– mice (PDF 723 kb)
ESM Fig. 2
Expression of GSK3B in the brain, hypothalamus, adipose tissue, liver, skeletal muscle and heart of RIP-Cre control and β-Gsk-3β –/– mice. Protein lysates were subjected to western blot analysis using anti-total GSK3A/B and either anti-α-tubulin or anti-β-actin. One of three independent experiments is shown (PDF 171 kb)
ESM Fig. 3
a Body weights of RIP-Cre control and β-Gsk3β –/– mice at indicated ages (RIP-Cre control, n ≥ 15; β-Gsk-3β –/–, n ≥ 20). b Food intake was measured in 12-week-old RIP-Cre control (n = 5) and β-Gsk-3β –/– mice (n = 5). Results are mean ± SEM (PDF 310 kb)
ESM Fig. 4
Fasting blood glucose levels were measured after overnight fasting in RIP-Cre control mice on either a normal diet (ND) or a high-fat diet (HFD) (n = 5) and β-Gsk3β –/– mice on either a normal diet (ND) or a high-fat diet (HFD) (n = 5) at indicated time points of HFD feeding. Results are mean ± SEM. *p < 0.05 (PDF 298 kb)
ESM Fig. 5
a Results of an intraperitoneal glucose tolerance test (IPGTT) on a normal diet (ND) or high-fat diet (HFD) compared between RIP-Cre control and β-Gsk3β –/– mice. All mice were 24 weeks of age at the time of these tests, and HFD was initiated at 12 weeks of age. Glucose levels were assessed at the indicated time intervals. Results are mean ± SEM. b IPGTT results in a shown as area under the curve. Results are mean ± SEM. *p < 0.05, **p < 0.01. c Comparison of beta cell mass from male RIP-Cre control and β-Gsk-3β –/– mice on either a normal diet at 12 weeks (Fig. 3a) or a high-fat diet at 24 weeks (Fig. 6c) of age. Results are expressed as beta cell mass (mg). All values are mean ± SEM. *p < 0.05, **p < 0.01 (PDF 461 kb)
ESM Fig. 6
a Islets were isolated from 12-week-old male RIP-Cre control and β-Gsk-3β –/– mice and lysates were subjected to western blot analysis using antibodies to the indicated proteins. One of three independent experiments is shown. Densitometric data for active β-catenin and total β-catenin were obtained and active β-catenin was normalised for total β-catenin. Protein levels are shown as mean ± SEM. *p < 0.05. b Islets were isolated from 12-week-old male RIP-Cre control and β-Gsk3β –/– mice and lysates were subjected to western blot analysis using antibodies to the indicated proteins. One of five independent experiments is shown. Densitometric data on total p27kip1, p21cip1 were normalised for α-tubulin. Protein levels are shown as mean ± SEM (PDF 243 kb)
ESM Table 1
Primers used for genotyping (PDF 20.7 kb)
ESM Table 2
Antibodies used in the experiments (PDF 28.4 kb)
Rights and permissions
About this article
Cite this article
Liu, Y., Tanabe, K., Baronnier, D. et al. Conditional ablation of Gsk-3β in islet beta cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetologia 53, 2600–2610 (2010). https://doi.org/10.1007/s00125-010-1882-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1882-x