
Abstract
Aims/hypothesis
There is convincing evidence that the endothelin system contributes to diabetic nephropathy and cardiovascular disease. This study aimed to assess the effects of the non-peptidergic endothelin receptor A (ETA) antagonist avosentan in a mouse model of accelerated diabetic nephropathy and atherosclerosis in comparison with the ACE inhibitor, quinapril.
Methods
Apolipoprotein E (Apoe) knockout (KO) mice (n = 20 per group, five groups) were randomised to the following groups: non-diabetic controls and streptozotocin-induced diabetic animals gavaged daily for 20 weeks with placebo, avosentan (high dose: 30 mg/kg, or low dose: 10 mg/kg) or quinapril (given in drinking water, 30 mg/kg).
Results
BP was unchanged by avosentan treatment but decreased with quinapril treatment. Diabetes-associated albuminuria was significantly attenuated by high-dose avosentan after 10 and 20 weeks of treatment. Diabetic animals showed a decreased creatinine clearance, which was normalised by avosentan treatment. In diabetic mice, high-dose avosentan treatment significantly attenuated the glomerulosclerosis index, mesangial matrix accumulation, glomerular accumulation of the matrix proteins collagen IV, and renal expression of genes encoding connective tissue growth factor, vascular endothelial growth factor, transforming growth factor β and nuclear factor κB (p65 subunit). Furthermore, high-dose avosentan treatment was also associated with reduced expression of the genes for ETA, ETB and angiotensin receptor 1. The renoprotective effects of avosentan were comparable or superior to those observed with quinapril. High-dose avosentan also significantly attenuated diabetes-associated aortic atherosclerosis in Apoe KO mice and reduced macrophage infiltration and aortic nitrotyrosine expression.
Conclusions/interpretation
This study demonstrates that ETA blockade with avosentan may provide an alternate therapeutic strategy for the treatment of diabetic micro- and macrovascular complications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The vasoactive peptide endothelin-1 (ET-1) has been recognised as playing a role in progressive renal injury in diabetic nephropathy via its vasoactive, pro-fibrotic and inflammatory properties [1]. Transgenic mice over producing ET-1 develop glomerulosclerosis and interstitial fibrosis without concurrent hypertension, suggesting that elevation in ET-1 contributes to renal fibrosis [2]. While blockade of the renin–angiotensin system (RAS) remains the major therapeutic approach to retard renal injury and vascular disease in diabetes, progression to end-stage renal disease still occurs in 34–47% of type 1 diabetic patients [3] and further therapeutic strategies are required to reduce this rate.
Of the two endothelin receptors, ETA and ETB, ETA antagonism has shown beneficial results in animal models of diabetes, with treatment with LU 135252 or FR 139317 providing renoprotective effects in association with reduced chemokine and cytokine expression, as well as attenuation of various mediators of fibrosis and sclerosis [4–6].
In a streptozotocin-induced model of diabetes in the rat, studies using dual ETA/ETB antagonists have generally shown them to also be renoprotective in preventing proteinuria and reducing fibrotic markers such as collagen I, fibronectin and transforming growth factor β (TGF-β) and also to normalise glomerular matrix protein levels [1]. Furthermore, the dual ETA/ETB antagonist bosentan has been shown to attenuate the increase in mesangial matrix deposition and glomerular basement membrane thickening seen in diabetic rats [7]. Not all treatments with dual antagonists have been shown to be beneficial, however; in a rat model of ischaemic acute renal failure, ETA/ETB antagonism (SB 209670) was found to be detrimental, whereas ETA antagonism alone (SB 234551) was beneficial on renal injury variables [8]. Thus it is possible that selective ETA antagonism may be a preferable approach to optimise renoprotection.
The non-peptidergic orally active ETA antagonist, avosentan, has been shown to be well tolerated in healthy individuals [9]. Recent clinical studies showed that treatment with avosentan attenuated urinary albumin excretion in diabetic patients, particularly when treatment was given in addition to ACE inhibition, the standard treatment for diabetic nephropathy [10]. However, the mechanism responsible for this renoprotective effect remains unclear. Furthermore, it is not known whether ETA antagonism also confers macrovascular protection in diabetes.
Thus the aim of this study was to investigate the reno- and vasculoprotective effects of the novel orally active ETA antagonist avosentan in diabetic apolipoprotein E (Apoe) knockout (KO) mice and to compare these effects with those conferred by a well known end-organ protective agent in diabetes, the ACE inhibitor quinapril.
Methods
The diabetic Apoe KO mouse represents an animal model combining hyperglycaemia and hyperlipidaemia. This mouse model demonstrates not only accelerated renal injury, including increased albuminuria, glomerulosclerosis and interstitial matrix accumulation [11], but also diabetes-associated atherosclerosis [12]. Six-week-old homozygous Apoe KO male mice (back-crossed 20 times from the C57BL/6 strain; Animal Resource Centre, Canning Vale, WA, Australia) were housed at the Precinct Animal Centre of the Baker IDI Heart and Diabetes Research Institute and studied according to the principles devised by the Animal Welfare Committee of the Baker IDI Heart and Diabetes Research Institute and Alfred Hospital under guidelines laid down by the National Health and Medical Research Council of Australia. Apoe KO mice were rendered diabetic by five daily i.p. injections of streptozotocin (Sigma-Aldrich, St Louis, MO, USA) at a dose of 55 mg/kg in citrate buffer. Only animals with blood glucose levels >15 mmol/l 1 week after the induction of diabetes were included in the study. The animals had unrestricted access to water and feed and were maintained on a 12 h light–12 h darkness cycle in a non-pathogen-free environment on standard mouse chow (Specialty Feeds, Glen Forrest, WA, Australia). Following the induction of diabetes (>90% of injected mice), both the non-diabetic Apoe KO controls (n = 20) and diabetic animals were further randomised (n = 20 per group) to receive 20 weeks of treatment with placebo (acetate buffer alone) or avosentan (SPP-301; 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidin-4-yl]- amide, donated by Speedel Pharma, Basel, Switzerland, with a median inhibitory concentration (IC50) = 3 nmol/l and a ∼500-fold selectivity for ETA over ETB [13]). Avosentan was given as either a low dose (10 mg/kg) or high dose (30 mg/kg; both doses dissolved in acetate buffer) treatment. Avosentan and placebo buffer were administered daily by gavage. Another two groups of mice consisting of non-diabetic and diabetic animals received quinapril in drinking water (30 mg/kg) for 20 weeks. Data from avosentan and quinapril-treated control animals are not presented.
Systolic BP was assessed by a non-invasive tail cuff system in conscious mice at the end of the study, as described [14]. Animals were habituated to the device before formal measurements began.
After 20 weeks, the animals were anaesthetised by an i.p. injection of sodium pentobarbitone (100 mg/kg body weight; Euthatal, Sigma-Aldrich, Castle Hill, NSW, Australia). Blood was collected from the left ventricle, centrifuged and plasma and remaining blood cells were stored at −20°C and at 4°C, respectively. Subsequent analysis of glycated haemoglobin levels was determined in lysates of erythrocytes by HPLC (Biorad, Richmond, CA, USA). Total cholesterol and triglyceride concentrations were measured in plasma with a standard commercial enzymatic assay using a Beckman Coulter LX20PRO Analyser (Cat No. 467825 Beckman Coulter Diagnostics, Gladesville, NSW, Australia). Kidneys and aortas were rapidly dissected and kidneys were weighed (wet weight) before both organs were either snap frozen in liquid nitrogen and stored at −80°C, or put in buffered formalin (10%, vol./vol.) before being embedded in paraffin for later immunohistochemical studies.
Assessment of renal function
At weeks 10 and 20, animals were housed for 24 h in metabolism cages for the collection of urine for the subsequent measurement of urinary excretion of albumin, and also food and water consumption (week 20 only).
Urinary albumin excretion was measured by a mouse albumin ELISA kit, according to the kit protocol (Bethyl Laboratories, Montgomery, TX, USA). Urinary and serum creatinine concentrations were measured by HPLC, according to the Animal Models of Diabetic Complications Consortium (AMDCC) guidelines (www.amdcc.org/shared/showFile.aspx?doctypeid=3&docid=21, accessed 7 September 2009). Briefly, aliquots of urine and plasma were mixed with acetyl nitrile (sample 1:5) and centrifuged at 4°C. The supernatant fraction was then removed, dried in a Speedivac (Thermo electron; Biolabs, Clayton, VIC, Australia) and resuspended in 10 mmol/l ammonium acetate, pH 3.2. Samples were then injected into a C18 column (Waters Division of Millipore, Marlborough, MA, USA) and detected at 235 nm using a Hewlett Packard PDA detector (1100 Series, Hewlett Packard, Singapore). Creatinine clearance was estimated as the ratio of daily urinary creatinine excretion to plasma creatinine concentration, and expressed as ml min−1 m−2 surface area.
Evaluation of renal structural injury
Mesangial area
Kidney sections (3 µm) were stained with periodic acid–Schiff (PAS). Mesangial area was analysed (per cent glomerular area) from digital pictures of glomeruli (15 glomeruli per kidney per animal) using Image-Pro Plus v6.0 (Media Cybernetics, Bethesda, MD, USA).
Glomerulosclerosis
The glomerulosclerosis index (GSI) was calculated as reported previously [11]. Briefly, this semi-quantitative evaluation of the degree of glomerular injury (25 glomeruli per kidney) included assessment of mesangial matrix accumulation and cellularity in the glomeruli tuft, with scoring being conducted in a blinded manner.
Assessment of plaque area
The en face analysis of atherosclerotic lesions was conducted as described previously [15], being stained with Sudan IV-Herxheimer’s solution (BDH, Poole, UK), and photographs of opened aortas digitised using a dissecting microscope (Olympus SZX9; Olympus Optical, Tokyo, Japan) and a digital camera (Axiocam colour camera; Carl Zeiss, North Ryde, NSW, Australia), with plaque area calculated as the proportion of aortic intimal surface area occupied by red-stained plaque (Adobe Photoshop v 6.0.1; Adobe Systems, Chatswood, NSW, Australia).
Immunohistochemistry
Paraffin sections (4 μm thick) of kidneys and aorta were used to stain for collagen type IV (goat polyclonal; Southern Biotechnology, Birmingham, AL, USA; diluted 1:500), fibronectin (rabbit polyclonal; Dako Cytomation, Glostrup, Denmark; diluted 1:800), nitrotyrosine (rabbit polyclonal; Millipore, Billerica, MA, USA; 1:40) or F4/80 (rat monoclonal; Abcam, Cambridge, MA, USA; 1:50). In brief, sections were dewaxed, hydrated, and quenched with 3% H2O2 in TRIS-buffered saline (pH 7.6) to inhibit endogenous peroxidase activity. Sections to be incubated with collagen IV primary antibody were digested with 0.4% pepsin (Sigma-Aldrich, Castle Hill, NSW, Australia) in 0.01 mol/l HCl at 37°C for 10 min, followed by 0.5% skim-milk for 15 min. Sections were then incubated with Protein Blocking Agent (Thermo Electron, Pittsburgh, PA, USA) for 60 min at room temperature, then in primary antibody overnight at 4°C in a humidified chamber. Sections incubated with the anti-collagen IV antibody then had excess avidin/biotin blocked (Avidin-Biotin blocking kit, Vector Laboratories, Burlingame, CA, USA). Both sets of sections were then incubated in the appropriate biotinylated secondary antibody (all from Vector Laboratories), followed by horseradish peroxidase-conjugated streptavidin (VECTASTAIN Elite ABC Staining Kit; Vector Laboratories). Sections for F4/80 detection were also amplified further (Dako Catalysed Signal Amplification Kit, according to instructions; Dako Cytomation). All sections were visualised with 3,3′-diaminobenzidine tetrahydrochloride/H2O2 (DAB; Sigma-Aldrich, St Louis, MO, USA). Finally, sections were counterstained with Mayer’s haematoxylin, dehydrated and coverslipped. All sections were examined under light microscopy (Olympus BX-50; Olympus Optical) and digitised with a high-resolution camera. All digital quantification (Image-Pro Plus, v6.0) and assessments were performed in a blinded manner.
Quantitative RT-PCR
Total RNA was extracted after homogenising one quarter of a whole kidney (Polytron PT-MR2100; Kinematica, Littau/Lucerne, Switzerland) in TRIzol Reagent (Invitrogen Australia, Mt Waverly, VIC, Australia). Contaminating DNA was removed after treatment with DNA-free DNAse according to the manufacturer’s specifications (Ambion, Austin, TX, USA). Finally, DNA-free RNA was reverse transcribed into cDNA using the Superscript First Strand Synthesis System (Life Technologies BRL, Grand Island, NY, USA).
Expression of genes encoding connective tissue growth factor (CTGF), vascular endothelial growth factor (VEGF), fibronectin, collagen IV (α1), α-smooth muscle actin (α-SMA), TGF-β, TGF-β receptor II, angiotensin receptors AT1a and AT1b, ETA and ETB, the p65 subunit of nuclear factor κB (NF-κB) and osteopontin were analysed by quantitative RT-PCR (qRT-PCR) using the Taqman system based on real-time detection of accumulated fluorescence (ABI Prism 7500; Perkin-Elmer, Foster City, CA, USA); probe and primer sequences are shown in Electronic supplementary material (ESM) Table 1. Fluorescence for each cycle was analysed quantitatively and gene expression normalised relative to the expression of the housekeeping gene for 18S rRNA (18S ribosomal RNA Taqman Control Reagent kit), which was multiplexed together with the gene of interest. Probes and primers were designed using a Primer Express program and were purchased from Applied Biosystems (ABI, Foster City, CA, USA). Amplifications were performed with the following time course: 50°C for 2 min and 95°C for 10 min and 40 cycles of 94°C for 20 s and at 60°C for 1 min. Results were expressed relative to non-diabetic (control) placebo-treated Apoe KO mice, which were arbitrarily assigned a value of 1.
Statistical analysis
Data were analysed by ANOVA. Comparisons of group means were performed by Fisher’s least-significant different method. Analyses were performed using SPSS (SPSS v15.0). Data are shown as means ± SEM unless otherwise specified. p < 0.05 was considered statistically significant.
Results
Metabolic variables
Systolic BP
There was no change in systolic BP when comparing diabetic Apoe KO mice with non-diabetic Apoe KO after 20 weeks of diabetes. Neither 10 nor 30 mg/kg avosentan significantly influenced BP when compared with placebo-treated animals; however, animals treated with quinapril had significantly lower systolic BP after 20 weeks of treatment (Table 1).
Water intake and urinary volume
All diabetic animals demonstrated significantly increased fluid intake compared with their relative controls, regardless of treatment. Similarly, all diabetic animals had significantly higher urinary outputs, regardless of treatment (Table 1).
Plasma cholesterol, glucose and glycated haemoglobin levels
Levels of plasma glucose, glycated haemoglobin, total cholesterol and HDL- and LDL-cholesterol were all significantly higher in diabetic Apoe KO mice compared with non-diabetic controls, and were not affected by any of the treatments (Table 1).
Body and organ weights
As expected, all diabetic animals were significantly lighter than control animals at the end of the study (Table 1). No differences in body weight were found between treatment groups in either the control or diabetic groups. Diabetic animals showed significantly increased kidney:body weight ratio, suggesting renal hypertrophy. High-dose avosentan treatment tended to reduce kidney:body weight ratio compared with diabetic Apoe KO mice (p = 0.053) and with diabetic Apoe KO mice treated with low-dose avosentan (p < 0.05) (Table 2).
Renal function and structure
Urinary albumin levels and creatinine clearance
The 24 h urinary albumin excretion was significantly increased in diabetic Apoe KO mice compared with non-diabetic control Apoe KO mice after 10 and 20 weeks of diabetes and was significantly attenuated in diabetic animals treated with high-dose avosentan and quinapril (Fig. 1a, b) with both these treatments reducing urinary albumin excretion to a similar degree. Low-dose avosentan reduced albuminuria only at the week 20 time point.

a Twenty-four hour urinary albumin excretion from diabetic (D) and non-diabetic control (C) Apoe knockout mice after 10 weeks (a) and 20 weeks (b) of diabetes. Animals were treated with placebo (D and C), high-dose avosentan (H; 30 mg/kg), low-dose avosentan (L; 10 mg/kg) or quinapril (Q; 30 mg/kg). Avosentan (30 mg/kg) and quinapril treatment decreased urinary albumin excretion in diabetic Apoe KO mice at both time points, with 10 mg/kg avosentan significantly reducing excretion at 20 weeks. Data are expressed as geometric means ± tolerance factor for n = 16–19 mice at 10 weeks, n = 9–15 at 20 weeks. c Week 20 24 h creatinine clearance was decreased in diabetic animals, but was normalised by avosentan and quinapril treatment. n = 7–8 mice per group. p < 0.05: *vs D; †vs C; ‡vs D + L; §vs D + Q
Creatinine clearance was significantly reduced in placebo-treated diabetic Apoe KO mice compared with placebo-treated non-diabetic control mice, reflecting impairment in renal function (Fig. 1c). This impairment was not seen in any avosentan-treated diabetic groups.
GSI, mesangial area
There was a dose-dependent reduction in mesangial area in avosentan-treated diabetic mice, with high-dose avosentan mice having significantly attenuated mesangial expansion (Fig. 2a, b). Quinapril treatment did not reduce mesangial area after 20 weeks of treatment in diabetic Apoe KO mice. Diabetic Apoe KO mice had significantly more glomerulosclerosis compared with control animals as assessed by the GSI (Fig. 2c). High-dose avosentan and quinapril treatments significantly reduced GSI in diabetic animals, with both treatments reducing GSI to a value similar to that seen in non-diabetic control Apoe KO mice.

a PAS stained glomeruli; scale bar, 20 μm. Percentage mesangial area (b) and glomerulosclerosis index (GSI) (c) from PAS-stained Apoe KO mouse kidneys, both of which were significantly reduced by high-dose (30 mg/kg) avosentan treatment in diabetic mice; n = 8 animals per group. Group abbreviations: D, diabetic and C, non-diabetic control; H, high-dose avosentan treatment (30 mg/kg) and L, low-dose (10 mg/kg) avosentan treatment; Q, quinapril treatment (30 mg/kg). p < 0.05: *vs D; †vs C; §vs D + Q; ‡vs D + L. n = 8 per group for mesangial area
Mediators of fibrogenesis
By 20 weeks of treatment, there was a significant elevation in the expression of the gene encoding the NF-κB subunit p65 in the diabetic kidney (Table 3). Nevertheless, expression of the genes encoding the profibrotic mediators fibronectin, collagen IV, α-SMA, CTGF, TGF-β and TGF-β receptor II was decreased in diabetic mice treated with avosentan, with high-dose avosentan treatment generally being more effective than low-dose treatment.
Furthermore, the expression of certain genes, such as those for CTGF, VEGF, TGF-β and fibronectin, was reduced with high-dose avosentan treatment; however, this was not observed with quinapril treatment. Diabetes was associated with increased accumulation of collagen IV and fibronectin protein staining in glomeruli compared with untreated diabetic Apoe KO mice (Figs 3 and 4). High-dose avosentan treatment attenuated fibronectin and collagen IV accumulation in diabetic mice, and while quinapril significantly reduced collagen IV staining, it did not reduce fibronectin protein levels.

a Photomicrographs of Apoe KO mouse glomeruli stained for collagen IV; scale bar represents 20 μm. Collagen IV gene expression was significantly reduced by avosentan and quinapril treatment (b) (n = 6–10 per group). Digital quantification showed that the percentage area of glomeruli (c) stained positively for collagen IV was significantly reduced by high-dose (30 mg/kg) avosentan treatment (n = 8 per group). Group abbreviations: D, diabetic and C, non-diabetic control; H, high-dose avosentan treatment (30 mg/kg) and L, low-dose (10 mg/kg) avosentan treatment; Q, quinapril treatment (30 mg/kg). p < 0.05: *vs D; †vs C; §vs D + Q; ‡vs D + L

a Photomicrographs of Apoe KO mouse glomeruli stained for fibronectin; scale bar represents 20 μm. Fibronectin gene expression was significantly reduced by high-dose avosentan treatment (b) (n = 6–10 per group).Digital quantification of the percentage area of glomeruli (c) stained positively for fibronectin (n = 8 per group). Group abbreviations: D, diabetic and C, non-diabetic control; H, high-dose avosentan treatment (30 mg/kg) and L, low-dose (10 mg/kg) avosentan treatment; Q, quinapril treatment (30 mg/kg). p < 0.05: *vs D; †vs C; §vs D + Q; ‡vs D + L
High-dose avosentan treatment was also associated with reduced gene expression of the pro-inflammatory mediators VEGF, the p65 subunit of NF-κB and osteopontin, as well as reduced levels of both endothelin receptors. Quinapril significantly reduced At1b (also known as Agtr1b), collagen IV, α-SMA (also known as Acta2) and p65 (also known as Rela) gene expression in diabetic mice, and tended to reduce Ctgf gene expression (p = 0.068) but, unlike high-dose avosentan treatment, quinapril had no significant effect on expression of genes encoding AT1a receptor, VEGF, TGF-β, TGF-β II receptor or the endothelin receptors.
Anti-atherosclerotic effects conferred by avosentan
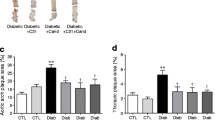
As previously published by our group [16], the induction of diabetes in Apoe KO mice resulted in an increase in atherosclerotic plaque area in the total aorta, comprising arch, thoracic and abdominal aorta, as well as in these individual regions of the aorta (Fig. 5). There was a dose-dependent attenuation of plaque area with avosentan treatment, with high-dose avosentan significantly reducing plaque area by approximately 40% in the total aorta, as well as ∼45% reduction in the thoracic aorta. Quinapril treatment significantly reduced plaque area in all parts of the aorta to levels similar to those observed in non-diabetic Apoe KO mice, and overall was more effective than avosentan treatment in reducing plaque accumulation.

a Photomicrographs of en face mouse aorta stained with Sudan IV. Atherosclerotic plaque is stained red with Sudan IV in (left to right) aortic arch, thoracic and abdominal aorta. Percentage of plaque area in the total aorta (b), the aortic arch (c), the thoracic aorta (d) or the abdominal aorta (e) in diabetic and control Apoe KO mice with and without avosentan treatment (n = 8–10 per group). High-dose avosentan treatment significantly reduced aortic plaque area in diabetic mice, the predominant reductions being seen in the thoracic aorta. As expected, quinapril treatment significantly decreased diabetic plaque area in all areas of the aorta. Group abbreviations: D, diabetic and C, non-diabetic control; H, high-dose avosentan treatment (30 mg/kg) and L, low-dose (10 mg/kg) avosentan treatment; Q, quinapril treatment (30 mg/kg). p < 0.05: *vs D; †vs C; §vs D + Q
There was a significant increase in staining for the macrophage marker, F4/80, in diabetic Apoe aorta (Fig. 6) reflecting an increase in macrophage infiltration in the aorta of these diabetic Apoe KO mice. This was decreased to control levels by both avosentan and quinapril treatment, suggesting a reduction in macrophage infiltration by both these treatments. To assess the level of oxidative stress in the aorta, aortas were stained for nitrotyrosine, a product of peroxynitrite-mediated protein oxidation (Fig. 7). In agreement with previous data from our group, diabetes was associated with a significant increase in nitrotyrosine staining [17], with this being significantly reduced by treatment with avosentan and quinapril. Indeed, both treatments reduced levels to those seen in aortas from non-diabetic Apoe KO mice.

a Photomicrographs of Apoe KO mouse aorta stained for the macrophage marker, F4/80. Scale bar, 100 μm. b Digital quantification of the percentage of aortic area stained, excluding the adventitia (n = 7–8 per group). F4/80 significantly increased in the aorta of diabetic mice compared with non-diabetic controls, with all treatments decreasing staining back to control levels. Group abbreviations: D, diabetic and C, non-diabetic control; H, high-dose avosentan treatment (30 mg/kg) and L, low-dose (10 mg/kg) avosentan treatment; Q, quinapril treatment (30 mg/kg). p < 0.05: *vs D; †vs C

a Photomicrographs of Apoe KO mouse aorta stained for nitrotyrosine (scale bar, 100 μm). b Digital quantification of the percentage of aortic area stained, excluding the adventitia. Nitrotyrosine significantly increased in the aorta of diabetic mice compared with non-diabetic controls, with all treatments decreasing nitrotyrosine staining back to control levels (n = 7–8 per group). Group abbreviations: D, diabetic and C, non-diabetic control; H, high-dose avosentan treatment (30 mg/kg) and L, low-dose avosentan treatment; Q, quinapril treatment (30 mg/kg). p < 0.05: *vs D; †vs C
Discussion
Diabetes is associated with significant changes in the expression and regulation of the endothelin system. In this study, treatment with the selective ETA antagonist, avosentan, significantly attenuated functional and structural renal injury in diabetic Apoe KO mice, associated with reduced accumulation of extracellular matrix proteins such as collagen IV. Avosentan also attenuated atherosclerotic plaque area in a dose-dependent manner. These beneficial effects were observed despite BP, lipid and glucose levels remaining unchanged by avosentan treatment. These data indicate that ETA plays a key role in the development and progression of diabetic complications, with avosentan treatment providing both reno- and vasculoprotection.
There is increasing evidence to suggest there is altered expression and distribution of various components of the endothelin system in diabetic nephropathy [18, 19]. The mechanism by which ET-1 promotes renal and vascular injury in diabetes is an area of active and ongoing investigation. Certainly, ET-1 has important haemodynamic effects that reduce renal blood flow and glomerular filtration at concentrations that do not affect BP. ET-1 is known to induce glomerular capillary hypertension and increase glomerular permeability [20]. In our study, increased albuminuria and reduced creatinine clearance were associated with diabetes in the Apoe KO mouse [21], with both being attenuated by avosentan in a dose-dependent fashion. However, these improvements may also reflect the range of non-haemodynamic actions of ET-1, which impact on pathways leading to cellular proliferation, inflammation and fibrogenesis.
It is predicted that the increase in ET-1 observed in diabetes would enhance renal injury by directly influencing pathways that have previously been shown, albeit predominantly in in vitro studies, to be activated by ET-1 [1]. This includes NF-κB, which activates expression of key pro-inflammatory products that contribute to diabetic kidney disease [22]. In our study, expression of the gene encoding the p65 subunit of NF-κB was significantly increased in the diabetic kidney and was significantly attenuated by ETA antagonism with avosentan.
The diabetic Apoe KO mouse represents a model of advanced diabetic glomerular injury. Interventions used in the clinical setting, such as ACE inhibitors and angiotensin receptor blockade, have been shown to be renoprotective and anti-atherosclerotic in this model [15, 23]. Treatment with high-dose avosentan for 20 weeks resulted in a significant reduction in GSI and mesangial area when compared with placebo-treated diabetic Apoe KO mice, with levels of sclerotic markers such as fibronectin and collagen IV being reduced in avosentan-treated diabetic animals. Indeed, ETA blockade was more effective than quinapril in reducing a range of profibrotic mediators and markers, such as expression of the genes encoding CTGF and both gene expression and protein production of collagen IV.
Interestingly, we found that ETA blockade but not ACE inhibition reduced mesangial area in diabetic mice. Although it has previously been reported that mesangial damage in human diabetic nephropathy is clearly linked to progression of disease [24], the failure of ACE inhibition to reduce matrix accumulation is consistent with previous studies by our group, which suggest that this morphological variable is not readily influenced by blockade of the RAS [25]. Interestingly, a recent clinical report in type 1 diabetic patients has failed to demonstrate a significant decrease in mesangial expansion with ACE inhibitor treatment [26]. These data do not exclude a role for ACE inhibition in decreasing renal structural injury in diabetic mice, as in this study we found that quinapril treatment significantly decreased other markers of renal damage, including renal collagen IV.
Consistent with the renal structural and functional improvements observed with avosentan treatment in diabetic Apoe KO mice, avosentan treatment was associated with decreased renal expression of genes for a number of growth factors which have been implicated in the pathogenesis of diabetic nephropathy. For example, VEGF is closely linked to the increased glomerular permeability seen in diabetes, and this appears to be partly mediated by the AT1 receptor subtype [27]. Furthermore, the pro-sclerotic growth factor CTGF has been reported to play a role in renal fibrosis in diabetic Apoe KO mice [28]. High-dose avosentan treatment decreased renal expression of genes encoding CTGF and VEGF in diabetic Apoe KO mice, consistent with the beneficial effects on renal fibrosis and reduced albuminuria observed with avosentan treatment. Furthermore, expression of the genes encoding the pro-sclerotic TGF-β and the TGF-β receptor II were significantly attenuated by ET receptor antagonism. Similarly, expression of the gene encoding osteopontin was decreased in diabetic mice treated with avosentan. Osteopontin has been identified as a marker of mesangial expansion in the diabetic mouse kidney [29], the severity of diabetic nephropathy in patients [30] and development of atherosclerosis [31]. The decrease in osteopontin expression with avosentan treatment is therefore consistent with decreases in the expression of genes encoding AT1a, TGF-β and NF-κB p65 subunit, which stimulate and are stimulated by osteopontin [32].
In the present study, treatment with high-dose avosentan significantly decreased expression of genes for both endothelin receptor subtypes. This is consistent with previous results showing that the ETA/ETB antagonist bosentan was associated with decreased ETA and ETB levels in diabetic rats [7].
Furthermore, avosentan treatment significantly reduced expression of the gene encoding the AT1a receptor, whereas quinapril did not have an effect on this receptor. It has been shown in vitro that ET1 and angiotensin II (Ang II) may interact [33]. The ETA receptor antagonist, BQ-123, was shown to decrease Ang II-stimulated fibronectin production in mesangial cells, as well as decreasing cell proliferation [34]. Thus, in addition to ET1 increasing pro-sclerotic factors in the kidney directly [35], avosentan may be reducing fibrosis via a reduction in the AT1 receptor, which itself has been shown to confer profibrotic effects in the kidney in diabetes [36].
Consistent with human studies of avosentan treatment [10], systolic BP was unchanged in diabetic Apoe KO mice after 20 weeks of treatment with either dose. This contrasts with some other ETA antagonists, which have been linked to changes in BP (BQ-123 [37, 38] and ABT-627 [39]), although not all ETA blockers have been found to affect BP (e.g. LU 135252 [6, 40, 41], TBC 3214 [42] and FR 139317 [4]). Therefore, the renoprotective effects observed with avosentan treatment in this study were independent of effects on BP. In contrast, quinapril treatment for 20 weeks was associated with a significant reduction in BP. Indeed, it is likely that the antihypertensive effect observed with quinapril treatment contributed at least in part to the renoprotective effects observed with this drug.
As well as being an excellent model of renal injury associated with diabetes, the diabetic Apoe KO mouse is widely regarded as the best model to assess accelerated atherosclerosis associated with diabetes. The vascular lesions that occur mimic those of human atherosclerosis and range from early lesions to advanced fibro-fatty plaques in the aorta [43]. We demonstrate that selective ETA antagonism significantly reduces aortic plaque formation in this model, independently of effects on BP, lipid or glucose levels. This anti-atherosclerotic effect of avosentan was associated with a significant reduction in macrophage infiltration and reduced nitrotyrosine levels, reflecting a decrease in oxidative stress. However, in contrast to the effects on renal variables, ACE inhibition was more effective in prevention of plaque formation compared with avosentan, possibly because of the added benefit of BP reduction with quinapril. These findings indicate that in addition to the renoprotective actions of avosentan, ETA blockade also has the potential to reduce macrophage infiltration and macrovascular oxidative stress, mechanisms considered to play vital roles in the development of diabetes-associated atherosclerosis.
In summary, the novel orally active ETA receptor antagonist avosentan significantly attenuated renal functional and structural injury in a mouse model of accelerated diabetic nephropathy, the streptozotocin-induced diabetic Apoe KO mouse. The anti-albuminuric effect of avosentan was similar to that observed with quinapril, but unlike quinapril, it did not lower BP. Avosentan treatment was associated with a dose-dependent attenuation of renal fibrosis and a reduction of various pro-sclerotic growth factors. Therefore, avosentan may represent an alternative approach to RAS blockade for the treatment of diabetic nephropathy, or may be an appropriate agent to combine with ACE inhibitor treatment. Furthermore, since avosentan may provide concomitant protection against macrovascular disease in diabetes, such a treatment could simultaneously confer benefits against micro- and macrovascular disease in diabetes, potentially slowing progression of the two major causes of morbidity and mortality in diabetes, chronic kidney and cardiovascular disease.
Abbreviations
- α-SMA:
-
α-Smooth muscle actin
- AT1:
-
Angiotensin receptor 1
- CTGF:
-
Connective tissue growth factor
- ET-1:
-
Endothelin-1
- ETA:
-
Endothelin receptor A
- ETB:
-
Endothelin receptor B
- GSI:
-
Glomerulosclerosis index
- KO:
-
Knockout
- NF-κB:
-
Nuclear factor κB
- PAS:
-
Periodic acid–Schiff
- qRT-PCR:
-
Quantitative RT-PCR
- RAS:
-
Renin–angiotensin system
- TGF-β:
-
Transforming growth factor β
- VEGF:
-
Vascular endothelial growth factor
References
Neuhofer W, Pittrow D (2006) Role of endothelin and endothelin receptor antagonists in renal disease. Eur J Clin Invest 36(Suppl 3):78–88
Hocher B, Thone-Reineke C, Rohmeiss P et al (1997) Endothelin-1 transgenic mice develop glomerulosclerosis, interstitial fibrosis, and renal cysts but not hypertension. J Clin Invest 99:1380–1389
Warram JH, Scott LJ, Hanna LS et al (2000) Progression of microalbuminuria to proteinuria in type 1 diabetes: nonlinear relationship with hyperglycemia. Diabetes 49:94–100
Nakamura T, Ebihara I, Fukui M, Tomino Y, Koide H (1995) Effect of a specific endothelin receptor A antagonist on mRNA levels for extracellular matrix components and growth factors in diabetic glomeruli. Diabetes 44:895–899
Hocher B, Schwarz A, Reinbacher D et al (2001) Effects of endothelin receptor antagonists on the progression of diabetic nephropathy. Nephron 87:161–169
Dhein S, Hochreuther S, Aus Dem Spring C, Bollig K, Hufnagel C, Raschack M (2000) Long-term effects of the endothelin(A) receptor antagonist LU 135252 and the angiotensin-converting enzyme inhibitor trandolapril on diabetic angiopathy and nephropathy in a chronic type I diabetes mellitus rat model. J Pharmacol Exp Ther 293:351–359
Chen S, Evans T, Deng D, Cukiernik M, Chakrabarti S (2002) Hyperhexosemia induced functional and structural changes in the kidneys: role of endothelins. Nephron 90:86–94
Forbes JM, Hewitson TD, Becker GJ, Jones CL (2001) Simultaneous blockade of endothelin A and B receptors in ischemic acute renal failure is detrimental to long-term kidney function. Kidney Int 59:1333–1341
Dieterle W, Mann J, Kutz K (2004) Pharmacokinetics and pharmacodynamics of the ETA-selective endothelin receptor antagonist SPP301 in healthy human subjects. J Clin Pharmacol 44:59–66
Wenzel RR, Littke T, Kuranoff S et al (2009) Avosentan reduces albumin excretion in diabetics with macroalbuminuria. J Am Soc Nephrol 20:655–664
Lassila M, Seah KK, Allen TJ et al (2004) Accelerated nephropathy in diabetic apolipoprotein E-knockout mouse: role of advanced glycation end products. J Am Soc Nephrol 15:2125–2138
Tse J, Martin-McNaulty B, Halks-Miller M et al (1999) Accelerated atherosclerosis and premature calcified cartilaginous metaplasia in the aorta of diabetic male Apo E knockout mice can be prevented by chronic treatment with 17 beta-estradiol. Atherosclerosis 144:303–313
Battistini B, Berthiaume N, Kelland NF, Webb DJ, Kohan DE (2006) Profile of past and current clinical trials involving endothelin receptor antagonists: the NOVEL ‘Sentan’ class of drug. Exp Biol Med (Maywood) 231:653–695
Krege JH, Hodgin JB, Hagaman JR, Smithies O (1995) A noninvasive computerized tail–cuff system for measuring blood pressure in mice. Hypertension 25:1111–1115
Candido R, Jandeleit-Dahm KA, Cao Z et al (2002) Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 106:246–253
Candido R, Allen TJ, Lassila M et al (2004) Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation 109:1536–1542
Soro-Paavonen A, Watson AMD, Li J, et al (2008) Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 57:2461–2469
Jandeleit-Dahm K, Allen TJ, Youssef S, Gilbert RE, Cooper ME (2000) Is there a role for endothelin antagonists in diabetic renal disease? Diabetes Obes Metab 2:15–24
Khan MA, Dashwood MR, Mumtaz FH, Thompson CS, Mikhailidis DP, Morgan RJ (1999) Upregulation of endothelin A receptor sites in the rabbit diabetic kidney: potential relevance to the early pathogenesis of diabetic nephropathy. Nephron 83:261–267
King AJ, Brenner BM, Anderson S (1989) Endothelin: a potent renal and systemic vasoconstrictor peptide. Am J Physiol Renal Physiol 256:F1051–F1058
Calkin AC, Giunti S, Jandeleit-Dahm KA, Allen TJ, Cooper ME, Thomas MC (2006) PPAR-alpha and -gamma agonists attenuate diabetic kidney disease in the apolipoprotein E knockout mouse. Nephrol Dial Transplant 21:2399–2405
Gomez-Garre D, Largo R, Tejera N, Fortes J, Manzarbeitia F, Egido J (2001) Activation of NF-kappaB in tubular epithelial cells of rats with intense proteinuria: role of angiotensin II and endothelin-1. Hypertension 37:1171–1178
Jandeleit-Dahm K, Lassila M, Davis BJ et al (2005) Anti-atherosclerotic and renoprotective effects of combined angiotensin-converting enzyme and neutral endopeptidase inhibition in diabetic apolipoprotein E-knockout mice. J Hypertens 23:2071–2082
Steffes MW, Bilous RW, Sutherland DER, Mauer SM (1992) Cell and matrix components of the glomerular mesangium in type I diabetes. Diabetes 41:679–684
Cooper ME, Allen TJ, Macmillan PA, Clarke BE, Jerums G, Doyle AE (1989) Enalapril retards glomerular basement membrane thickening and albuminuria in the diabetic rat. Diabetologia 32:326–328
Mauer M, Zinman B, Gardiner R et al (2009) Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med 361:40–51
Rizkalla B, Forbes JM, Cao Z, Boner G, Cooper ME (2005) Temporal renal expression of angiogenic growth factors and their receptors in experimental diabetes: role of the renin–angiotensin system. J Hypertens 23:153–164
Lassila M, Jandeleit-Dahm K, Seah KK et al (2005) Imatinib attenuates diabetic nephropathy in apolipoprotein E-knockout mice. J Am Soc Nephrol 16:363–373
Susztak K, Bottinger E, Novetsky A et al (2004) Molecular profiling of diabetic mouse kidney reveals novel genes linked to glomerular disease. Diabetes 53:784–794
Yamaguchi H, Igarashi M, Hirata A et al (2004) Progression of diabetic nephropathy enhances the plasma osteopontin level in type 2 diabetic patients. Endocr J 51:499–504
Bruemmer D, Collins AR, Noh G et al (2003) Angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest 112:1318–1331
Lorenzen J, Shah R, Biser A et al (2008) The role of osteopontin in the development of albuminuria. J Am Soc Nephrol 19:884–890
Sorokin A, Kohan DE (2003) Physiology and pathology of endothelin-1 in renal mesangium. Am J Physiol Renal Physiol 285:F579–F589
Gomez-Garre D, Ruiz-Ortega M, Ortego M et al (1996) Effects and interactions of endothelin-1 and angiotensin II on matrix protein expression and synthesis and mesangial cell growth. Hypertension 27:885–892
Orth SR, Viedt C, Amann K, Ritz E (2001) Endothelin in renal diseases and cardiovascular remodeling in renal failure. Intern Med 40:285–291
Leehey DJ, Singh AK, Alavi N, Singh R (2000) Role of angiotensin II in diabetic nephropathy. Kidney Int Suppl 77:S93–S98
Goddard J, Johnston NR, Hand MF et al (2004) Endothelin-A receptor antagonism reduces blood pressure and increases renal blood flow in hypertensive patients with chronic renal failure: a comparison of selective and combined endothelin receptor blockade. Circulation 109:1186–1193
Spratt JC, Goddard J, Patel N, Strachan FE, Rankin AJ, Webb DJ (2001) Systemic ETA receptor antagonism with BQ-123 blocks ET-1 induced forearm vasoconstriction and decreases peripheral vascular resistance in healthy men. Br J Pharmacol 134:648–654
Sasser JM, Sullivan JC, Hobbs JL et al (2007) Endothelin A receptor blockade reduces diabetic renal injury via an anti-inflammatory mechanism. J Am Soc Nephrol 18:143–154
Barton M, Haudenschild CC, d’Uscio LV, Shaw S, Munter K, Luscher TF (1998) Endothelin ETA receptor blockade restores NO-mediated endothelial function and inhibits atherosclerosis in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A 95:14367–14372
Orth SR, Odoni G, Amann K, Strzelczyk P, Raschack M, Ritz E (1999) The ET(A) receptor blocker LU 135252 prevents chronic transplant nephropathy in the ‘Fisher to Lewis’ model. J Am Soc Nephrol 10:387–391
Fukuda G, Khan ZA, Barbin YP, Farhangkhoee H, Tilton RG, Chakrabarti S (2005) Endothelin-mediated remodeling in aortas of diabetic rats. Diabetes Metab Res Rev 21:367–375
Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R (1994) ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb 14:133–140
Jandeleit-Dahm K, Watson A, Soro-Paavonen A et al (2008) The novel endothelin receptor A (ET-A) antagonist SPP 301 attenuates albuminuria and renal structural injury in streptozotocin-induced diabetic ApoE knockout mice. J Hypertens 26:S194 abstract
Acknowledgements
A. M. D. Watson is currently supported by a National Health and Medical Research Council of Australia (NHMRC) Australian Biomedical Fellowship. T. J. Allen and K. A. M. Jandeleit-Dahm are supported by NHMRC Senior Research Fellowships. This study was also supported by the Diabetes Australia Research Trust (DART) and the Juvenile Diabetes Research Foundation (JDRF). This work has been published in abstract form [44].
Duality of interest
This work was partially supported by a grant-in-aid from Speedel Pharma (Basel, Switzerland).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 19 kb)
Rights and permissions
About this article
Cite this article
Watson, A.M.D., Li, J., Schumacher, C. et al. The endothelin receptor antagonist avosentan ameliorates nephropathy and atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia 53, 192–203 (2010). https://doi.org/10.1007/s00125-009-1540-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1540-3