
Abstract
Aims/hypothesis
GW501516, an agonist of peroxisome proliferator-activated receptor-δ (PPAR-δ), increases lipid combustion and exerts antidiabetic action in animals, effects which are attributed mainly to direct effects on skeletal muscle. We explored such actions further in isolated rat skeletal muscle.
Materials and methods
Specimens of rat skeletal muscle were pretreated with GW501516 (0.01–30 μmol/l) for 0.5, 4 or 24 h and rates of fuel metabolism were then measured. In addition, effects on mitochondrial function were determined in isolated rat liver mitochondria.
Results
At concentrations between 0.01 and 1 μmol/l, GW501516 dose-dependently increased fatty acid oxidation but reduced glucose utilisation in isolated muscle. Thus after 24 h of preincubation with 1 μmol/l GW501516, palmitate oxidation increased by +46±10%, and the following decreased as specified: glucose oxidation −46±8%, glycogen synthesis −42±6%, lactate release −20±2%, glucose transport −15±6% (all p<0.05). Reduction of glucose utilisation persisted independently of insulin stimulation or muscle fibre type, but depended on fatty acid availability (the effect on glucose transport in the absence of fatty acids was an increase of 30±9%, p<0.01), suggesting a role for the glucose–fatty acid cycle. At higher concentrations, GW501516 uncoupled oxidative phosphorylation by direct action on isolated mitochondria.
Conclusions/interpretation
GW501516-induced activation of PPAR-δ reduces glucose utilisation by skeletal muscle through a switch in mitochondrial substrate preference from carbohydrate to lipid. High concentrations of GW501516 induce mitochondrial uncoupling independently of PPAR-δ.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Peroxisome proliferator-activated receptors (PPARs) are regulators of fuel metabolism that mediate their actions by modulating gene expression. Agonists of the subtypes PPAR-α (fibrates) and PPAR-γ (thiazolidinediones) are in clinical use for the treatment of deranged lipid metabolism and type 2 diabetes. Their regulatory roles in fuel metabolism have been studied extensively [1, 2] and more recently PPAR-δ (also referred to as PPAR-β) has also been recognised as an important regulator of fuel handling [1, 3]. Since activation of PPAR-δ by specific agonists or genetic manipulation ameliorates hyperglycaemia, insulin resistance and dyslipidaemia in animal models with obesity and type 2 diabetes [4–7], PPAR-δ is regarded as a promising new target in the treatment of metabolic disorders.
The beneficial effects of PPAR-δ on fuel metabolism are attributed to the increased expression of genes involved in lipid metabolism, uncoupling of oxidative phosphorylation, and mitochondrial biogenesis, which leads to elevated energy expenditure and fat dissipation [4, 5, 8, 9]. In this context, the quantitatively most important target organ seems to be skeletal muscle, which makes a major contribution to whole-body fuel oxidation and expresses PPAR-δ more abundantly than other PPAR subtypes [10]. Accordingly, targeted overexpression of PPAR-δ in mouse skeletal muscle distinctly elevates the number of red type-1-like fibres, characterised by high mitochondrial density and oxidative capacity [9, 11]. Furthermore, several studies have consistently shown that agonists of PPAR-δ markedly increase the rate of fatty acid oxidation by specimens of skeletal muscle both ex vivo and in vitro [4, 5, 10].
In contrast to several reports documenting PPAR-δ-induced gene expression and fatty acid oxidation in skeletal muscle, the accompanying changes in muscle glucose metabolism are largely unknown. To close this gap in knowledge, we thoroughly explored the direct effects of GW501516, a potent and selective PPAR-δ agonist [6, 12], on glucose handling by freshly isolated native specimens of rat skeletal muscle.
Material and methods
Rats
Male Sprague–Dawley rats were purchased from the breeding facilities of the Medical University of Vienna (Himberg, Austria). They were kept under an artificial 12-h light/12-h dark cycle at constant room temperature and, unless stated otherwise, had free access to a conventional laboratory diet and tap water. All experiments were performed according to local law and to the principles of good laboratory animal care.
Isolated muscle strips
Pretreatment period
Food, but not water, was withdrawn 3–4 h before 6- to 8-week-old rats were killed by cervical dislocation between 10.00 and 11.00 h. Immediately after killing, two longitudinal strips of soleus or extensor digitorum longus (EDL) muscle per leg were prepared, weighed (approximately 25 mg/strip) and tied under tension on stainless steel clips [13]. Four muscle strips were thus available per rat, allowing the paired examination of three concentrations of GW501516 (gift of Hoffmann-La Roche, Basel, Switzerland) along with an intra-individual control. Effects on fuel metabolism were initially studied at three concentrations at which GW501516 was expected to dose-dependently activate PPAR-δ (0.01, 0.1, and 1 μmol/l; referred to as low concentrations) [6, 12]. When a pilot experiment with higher concentrations raised further mechanistic questions, an additional set of three concentrations, at which PPAR-δ activation was expected to be maximal, was also examined (3, 10 and 30 μmol/l GW501516; referred to as high concentrations).
According to procedures employed earlier [14], muscles were immediately put into coated Erlenmeyer flasks and provided with Cell Culture Medium 199 (pH 7.35, 5.5 mmol/l glucose; Sigma, St Louis, MO, USA; catalogue no. M-4530). If not stated otherwise, the medium was supplemented with 20% vol/vol fetal bovine serum (FBS; HyClone, Logan, UT, USA), 0.3% wt/vol fatty acid-free BSA, 5 mmol/l HEPES, 300 μmol/l palmitate (dissolved in ethanol; final concentration 0.25% vol/vol), 25 000 U/l penicillin G, 25 mg/l streptomycin, and 0.2 mg/l ciprofloxacin. Depending on the specific experiment, GW501516, WY14643 (Sigma) and/or cycloheximide (Sigma) were dissolved in DMSO (Sigma) and added as indicated. The final concentration of DMSO was always the same as in the respective control experiment (0.1 or 0.2% vol/vol).
The flasks were placed into a shaking water bath (37°C; 130 cycles/min) for pretreatment periods of 0.5 h (one strip in 3 ml medium per flask), 4 or 24 h (three strips from different rats in 12 ml per flask). Insulin was not present during pretreatment. Throughout the whole experiment, an atmosphere of 95% O2/5% CO2 was provided within the flasks.
Measurement period
Immediately after pretreatment, muscles were transferred into identical medium (one strip in 3 ml per flask) additionally supplemented with trace amounts of d-[U–14C]glucose, d-[U–14C]palmitic acid, [U-14C]methionine or 2-deoxy-d-[2,6] glucose plus d-[U-14C]sucrose (all from Amersham, Amersham, UK), and, if not stated otherwise, with a maximally effective concentration of human insulin (Actrapid; Novo, Bagsvaerd, Denmark; 25 nmol/l after 0.5 h, 100 nmol/l after 4 and 24 h pretreatment periods). The measurement period lasted 1 h, after which muscles were quickly removed from the flasks, blotted, and frozen in liquid nitrogen.
Analyses
All analytical methods have been described in detail previously [14–16]. In short, rates of CO2 production from glucose or palmitate (referred to as glucose and palmitate oxidation) were calculated from the conversion of [14C]glucose or [14C]palmitate into 14CO2, which was trapped with a solution containing methanol and phenethylamine (1:1). For the measurement of glycogen storage, frozen muscle strips were lysed in 1 mol/l KOH at 70°C and the net rate of glucose incorporation into glycogen (referred to as glycogen synthesis) was calculated from the conversion of [14C]glucose into [14C]glycogen. The glycogen content prevailing at the end of the experiment was determined by degrading glycogen in the muscle lysate to glucose units with amyloglucosidase, followed by measurement of glucose with an enzymatic kit (Human, Taunusstein, Germany) [14, 15]. Rates of lactate release were calculated from lactate accumulated in the incubation medium as measured with the spectrophotometric lactate dehydrogenase method [14, 15]. Intracellular accumulation of 2-deoxy-d-[2,6]glucose (referred to as glucose transport) was determined using d-[U-14C]sucrose as a marker of extracellular space, and the net rate of methionine incorporation into protein (referred to as protein synthesis) was calculated from the conversion of [14C]methionine into [14C]protein using muscle strips lysed in 1 mol/l NaOH [15]. For the determination of the energy charge, muscle strips were extracted with 3 mol/l perchloric acid, and ATP and phosphocreatine were measured spectrophotometrically after neutralisation with KOH [16].
AMP-activated protein kinase activity
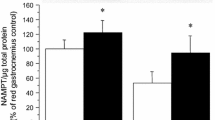
After 24 h of preincubation, specimens of EDL muscle were frozen in liquid nitrogen, crushed with a glass stirrer, and homogenised in Weinberg buffer (4°C). Lysates (approximately 0.1 g muscle/ml) were centrifuged and the protein concentration of the supernatant was measured with BSA as standard (BCA-protein assay; Pierce, Rockford, IL, USA). Aliquots were subjected to polyacrylamide gel electrophoresis under non-reducing conditions, transferred onto nitrocellulose membranes, blocked, exposed overnight at 4°C to the primary AMP-activated protein kinase (AMPK) antibody Anti-P-AMPK-α (rabbit polyclonal antibody, Thr172; Cell Signaling, Danvers, MA, USA), and exposed to the secondary antibody (horseradish peroxidase-conjugated donkey anti-rabbit immunoglobulin G; Amersham). Using the same blot, the procedure was then repeated with Anti-AMPK-α (Cell Signalling). The bands were visualised using the Super signal system from Pierce and Kodak XAR5-Omat films. The quotient of P-AMPK/AMPK is referred to as AMPK activity.
Isolated mitochondria
As described previously in more detail [14, 17], mitochondria were prepared from livers of 3-month-old rats killed by cervical dislocation. Mitochondria from liver rather than skeletal muscle were used, because they can be obtained in much higher yields and their behaviour is qualitatively identical with regard to the bioenergetic functions examined in this study. Livers were quickly excised and mitochondria were isolated in isolation buffer (0.25 mol/l sucrose, 20 mmol/l triethanolamine, 1 mmol/l EDTA; pH 7.4; 4°C) by tissue homogenisation and differential centrifugation. The protein content of the final mitochondrial suspension (30–40 g/l; kept at 4°C) was determined by the biuret method with BSA as standard.
Oxygen consumption was measured with a Clark-type oxygen electrode. Mitochondria (1 g/l protein) were preincubated for 3 min at 25°C in isolation buffer additionally containing 0.3% wt/vol BSA, the indicated concentration of GW501516, and 0.2% vol/vol DMSO. To stimulate mitochondrial respiration, 4 mmol/l inorganic phosphate was then added together with 5 mmol/l glutamate + 5 mmol/l malate (substrates for respiratory complex I) or 10 mmol/l succinate (substrate for complex II) + 2 μmol/l rotenone (blocker of complex I). After another 3 min, mitochondrial respiration was accelerated by the addition of 200 μmol/l ADP, allowing ATP synthesis, and the rates of oxygen consumption were measured in state 3 (i.e. in the presence of ADP). After the quantitative consumption of added ADP, we measured the rates of oxygen consumption in state 4 (i.e. in the absence of ADP). The energy-conserving capacity of mitochondria was determined as the respiratory control index (state 3/state 4). As a measure of the efficiency of mitochondrial ATP synthesis, the ratio of the total amount of ADP added per unit of oxygen consumed during state 3 respiration was calculated. All measurements were made in duplicate and intra-individual control values (same concentration of DMSO but no GW501516) were always determined in parallel.
Statistics
Because of the exploratory character of the study, statistics were used in a descriptive way. If not indicated otherwise, results are given as the mean±SEM and differences were analysed with two-tailed Student’s t-test (paired or unpaired, as appropriate) with p<0.05 considered significant.
Results
Dose- and time-dependent effects of GW501516 on muscle fuel handling
Effects of low concentrations of GW501516
Prolonged pretreatment of soleus muscle strips for 24 h with low concentrations of GW501516 (≤1 μmol/l) dose-dependently increased insulin-stimulated palmitate oxidation (Fig. 1a). This was accompanied by a general reduction in glucose utilisation, including distinct and dose-dependent decreases in glucose oxidation (Fig. 1b), glycogen content and synthesis (Fig. 1c,d), and lactate release (Fig. 1e). This switch in substrate preference occurred without any change in total protein synthesis and cellular energy charge (ATP, phosphocreatine; see electronic supplementary material [ESM] Table 3). AMPK activity was also unaffected by 1 μmol/l GW501516 (93±12% of intra-individual control; n=21; not significant).

Dose- and time-dependent effects of GW501516 on fuel metabolism in rat soleus muscle. Muscle strips were pretreated with GW501516 for 0.5, 4 or 24 h, before rates of palmitate oxidation (a), glucose oxidation (b), glycogen synthesis (d) and lactate release (e) were measured under insulin stimulation. Glycogen content (c) was determined at the end of the experiment. One out of four muscle specimens from the same rat served as a control (0 μmol/l GW501516), while three were exposed to low concentrations (0.01, 0.1 and 1 μmol/l; n=10–40 each) or high concentrations (3, 10, 30 μmol/l; n=6–12 each) of GW501516. Data are given as change vs intra-individual control (for control values in absolute terms see ESM Table 1). Data are mean±SEM. *p<0.05 vs control
In line with a PPAR-δ-mediated, hence genomic and delayed mechanism of action, no effects were observed after short-term pretreatment (0.5 or 4 h) with low concentrations of GW501516 (except for a moderate reduction of glycogen content after 4 h of pretreatment; Fig. 1). Dependence on a genomic mechanism was confirmed by the use of the protein synthesis inhibitor cycloheximide, which itself had a stimulatory effect on palmitate oxidation (p<0.01), but also abolished any effect of 24 h of exposure to 1 μmol/l GW501516 on glucose and palmitate oxidation (Fig. 2a,b).

Dependence of the effects of GW501516 on protein synthesis in rat soleus muscle. Muscle strips were pretreated with 1 mg/l of the protein synthesis inhibitor cycloheximide in combination with 1 μmol/l GW501516 for 24 h (a, b) or in combination with 30 μmol/l GW501516 for 0.5 h (c) before rates of glucose or palmitate oxidation were measured under insulin stimulation. Data are mean±SEM. n=6–12 for each comparison. *p<0.05 for presence (black bars) vs absence (white bars) of GW501516
Effects of high concentrations of GW501516
Twenty-four hours of pretreatment with high concentrations of GW501516 (≥3 μmol/l) further increased the rate of palmitate oxidation (Fig. 1a). Unlike low concentrations, however, high concentrations obviously activated a non-genomic mechanism of action, because (1) palmitate oxidation was elevated acutely after only 0.5 h of pretreatment (Fig. 1a) and (2) this rapid effect was insensitive to cycloheximide (Fig. 2c). The accompanying changes in glucose metabolism also differed from those induced by low concentrations, since pretreatment with 30 μmol/l GW501516 for 4 h stimulated glucose oxidation and pretreatment with ≥3 μmol/l GW501516 for 24 h induced a relative increase in glucose oxidation compared with the nadir seen at 1 μmol/l (1 vs 3, 10 and 30 μmol/l GW501516: p=0.045, p=0.055 and p=0.0025, respectively; Fig. 1b). Along with such an increase in substrate requirements for glucose oxidation, glycogen content decreased further in response to high concentrations of GW501516 (Fig. 1c), whereas net glycogen synthesis and lactate release were inhibited to extents similar to those seen at 1 μmol/l (Fig. 1d,e).
Genomic effects of GW501516 at low concentrations
Dependence on muscle fibre type
In white EDL muscle (Fig. 3) there was time-dependent impairment of insulin-stimulated glucose utilisation that was very similar to that seen in red soleus muscle (Fig. 1). The putatively PPAR-δ-mediated genomic effects of GW501516 were therefore independent of the muscle fibre type.

Dose- and time-dependent effects of GW501516 on glucose metabolism in rat extensor digitorum longus muscle. Muscle strips were pretreated with GW501516 for 0.5, 4 or 24 h, before rates of glucose oxidation (a), glycogen synthesis (c) and lactate release (d) were measured under insulin stimulation. Glycogen content (b) was determined at the end of the experiment. One out of four muscle specimens from the same rat served as a control (0 μmol/l GW501516), while three were exposed to the indicated concentrations of GW501516 (n=6–11 each). Data are given as changes vs intra-individual control (for control values in absolute terms see ESM Table 2). Data are mean±SEM. *p<0.05 vs control
Dependence on concomitant insulin stimulation
GW501516 impaired glucose metabolism both in the absence and in the presence of various concentrations of insulin (Fig. 4). In absolute terms, the GW501516-induced decrease in glucose utilisation was more distinct under insulin stimulation, whereas the relative decrease (%) was not affected by insulin (particularly obvious for glycogen synthesis; Fig. 4c).

Dependence of the effects of GW501516 on concomitant insulin stimulation. Rat soleus muscle strips were preincubated without GW501516 (white bars) or with 0.1 μmol/l GW501516 (black bars) for 24 h, before rates of glucose oxidation (a), lactate release (b) and glycogen synthesis (c) were measured under stimulation with 0, 1, 10 or 100 nmol/l insulin (as indicated). Glycogen content (d) was measured at the end of the experiment. Data are mean±SEM; n=9–14 each; *p<0.05, presence vs absence of GW501516. Values below x-axes: percent intra-individual decrease
Dependence on fatty acid availability
To clarify whether the GW501516-induced decrease in glucose utilisation depended on the availability of fatty acids, soleus muscle strips were incubated without fatty acids in the ambient medium during the whole experiment (FBS and palmitate omitted). Under these conditions, inhibition of glucose metabolism by 0.01 or 0.1 μmol/l GW501516 was the same as in the presence of extracellular fatty acids (compare Figs. 1 and 5), possibly because fatty acids were still available from intracellular lipid stores after 24 h of pretreatment. The distinct relative increase in glucose oxidation between 0.1 and 1 μmol/l GW501516 (p<0.001; Fig. 5a), however, suggests that accelerated depletion of intracellular lipid stores during preincubation with 1 μmol/l GW501516 had resulted in increased dependence on energy recruitment from glucose oxidation. In line with such a putative increase in carbohydrate requirements, the relative increase in glucose oxidation between 0.1 and 1 μmol/l GW510516 was accompanied by further glycogen depletion (Fig. 5b,c). Interdependence of fatty acid availability and cellular glucose utilisation is also shown by the divergent effects of 1 μmol/l GW501516 on glucose transport in the presence and absence of fatty acids (decrease and increase, respectively; Fig. 6).

Effects of GW501516 in the absence of fatty acids. Rat soleus muscle strips were pretreated with 0.01, 0.1 or 1 μmol/l GW501516 for 24 h without fatty acids in the ambient incubation medium, before rates of glucose oxidation (a) and glycogen synthesis (b) were measured under insulin stimulation. Glycogen content (c) was measured at the end of the experiment. Data are mean±SEM; n=17–18 each; *p<0.05 vs intra-individual control (0 μmol/l GW501516)

Effects of GW501516 on glucose transport in relation to fatty acid availability. Rat soleus muscle strips were pretreated with 0.01, 0.1 or 1 μmol/l GW501516 for 24 h in the presence (white bars) or absence (black bars) of fatty acids in the ambient incubation medium, before rates of radiolabelled 2-deoxy-glucose transport were measured under insulin stimulation. Data are given as changes vs the intra-individual control (0 μmol/l GW501516). Data are mean±SEM; n=6–12 each; *p<0.05 vs control
Dependence on PPAR subtype
Since agonists of PPAR-δ and PPAR-α have similar effects on the expression of lipid regulatory genes and fatty acid oxidation in cultured myocytes [10, 18], we examined the effects of the specific PPAR-α agonist WY14643 in the same protocol as that used with GW501516. At a range of concentrations that dose-dependently activates PPAR-α (0.1, 1 and 10 μmol/l) [19], WY14643 impaired glucose oxidation but had no further effects on glucose metabolism (Table 1).
Dependence on fuel selection by mitochondria
In all the above experiments, intracellular substrate stores were unlabelled, which implies that the measured oxidation rates could be influenced by changes in intracellular substrate cycling affecting the dilution of radiolabel in the intracellular substrate pool. To exclude misinterpretation, experiments were also performed with radiolabelled substrates provided throughout the complete 24 h pretreatment period (allowing for prelabelling of intracellular stores). Under these conditions, stimulation of palmitate oxidation and inhibition of glucose oxidation by 1 μmol/l GW5101516 persisted (palmitate oxidation, +21±9%, p=0.03; glucose oxidation, −28±4%, p<0.001), thus confirming that the described effects of GW501516 were, at least mainly, related to changes in mitochondrial substrate selection.
Non-genomic effects of GW501516 at high concentrations
Since agonists of other PPAR subtypes, α and γ, are known to affect mitochondrial function directly [14, 17], we also studied the effects of high concentrations of GW501516 on isolated mitochondria. Independently of whether substrates for respiratory complex I or II were provided, GW501516 dose-dependently increased state 4 respiration but reduced state 3 respiration, the respiratory control index and the ADP/O ratio (Fig. 7). This pattern of response clearly indicates uncoupling of oxidative phosphorylation and suggests that direct interaction with mitochondria could be responsible for the non-genomic stimulatory effect on both palmitate and glucose oxidation observed in soleus muscle exposed to high concentrations of GW501516 (Fig. 1).

Effects of GW501516 on isolated mitochondria. Isolated mitochondria from rat liver were treated with 10, 30 or 100 μmol/l GW501516 and rates of oxygen consumption in states 4 and 3 (i.e. in the presence and absence of ADP, respectively; a, b), the respiratory control index (ratio of oxygen consumption in state 3/state 4; c) and the ADP/O ratio (ADP/oxygen consumed during state 3 respiration; d) were determined in the presence of substrates for complex I (glutamate + malate) or complex II (succinate). Data are mean±SEM. n=6 each. *p<0.05 vs control (0 μmol/l GW501516)
Discussion
The study shows that the PPAR-δ agonist GW501516 has distinct direct effects on the glucose metabolism of rat skeletal muscle, adding to its established stimulatory impact on fatty acid oxidation [4, 5, 10]. Analysis of dependencies on exposure time and protein synthesis clearly suggests that GW501516 affects the fuel metabolism of isolated rat muscle via two independent mechanisms: (1) a delayed genomic mechanism sensitive to low concentrations of GW501516 and presumably due to the activation of PPAR-δ; and (2) a rapid non-genomic, and hence PPAR-independent, mechanism activated at higher concentrations and probably due to uncoupling of mitochondrial oxidative phosphorylation.
PPAR-δ-mediated actions
Genomic suppression of glucose utilisation by GW501516 is obviously mediated by PPAR-δ, because it occurs at concentrations reported to dose-dependently activate PPAR-δ in vitro and to circulate in GW501516-treated monkeys, in which changes in the plasma lipid profile are presumably mediated by PPAR-δ [6, 12]. As the impairment of glucose utilisation prevails independently of the muscle fibre type examined (i.e. in both red and white muscle), it is unlikely to depend on PPAR-δ-induced fibre type conversion which has been suggested to occur in mice subjected to overexpression of PPAR-δ in skeletal muscle [9, 11]. Furthermore, the finding that the PPAR-α agonist WY14643 likewise inhibits glucose utilisation in isolated muscle, but to a quantitatively much smaller extent than GW501516, corroborates previous evidence that these two PPAR subtypes transduce similar effects, but that PPAR-δ is of predominant importance in native skeletal muscle of rodents [5, 10].
The direct and marked impact of PPAR-δ on genes involved in lipid oxidation [5, 8, 18] suggests that the GW501516-induced increase in fatty acid utilisation could be the primary event, which gives rise to impairment of glucose utilisation via the well-known mutual inhibition of cellular glucose and fatty acid utilisation (glucose–fatty acid cycle) [20, 21]. This interpretation is compatible with our demonstration that GW501516-induced inhibition of glucose utilisation does not persist when cellular glucose requirements increase because of insufficient fatty acid availability or deteriorated mitochondrial ATP synthesis through the direct uncoupling of oxidative phosphorylation by high concentrations of GW501516. Furthermore, the parallel inhibition of all major pathways of cellular glucose metabolism is in agreement with the observed inhibitory effects of elevated fatty acid utilisation on the initial steps of muscle glucose metabolism, i.e. glucose transport and/or phosphorylation [20–22]. If GW501516-induced inhibition of glucose metabolism is thus due to a reversible shift in fuel selection, it is very understandable that such inhibition cannot be demonstrated in cells devoid of lipid fuel after prolonged incubation in fatty acid-free medium. Under such conditions, GW501516 even increases the rate of glucose transport (this study and [23]), presumably because activation of PPAR-δ increases carbohydrate requirements by accelerating the depletion of the remaining intracellular lipid stores and/or by triggering the expression of genes involved in mitochondrial uncoupling and energy dissipation [4, 5, 8, 18, 24]. Accordingly, GW501516 induces AMPK activation, a characteristic response to impaired energy availability, in cells devoid of lipid fuel [23], but not in cells supplied with fatty acids (this study). Any such interpretation casts doubt on the contention that GW501516 could exert antidiabetic action via a direct stimulatory effect on AMPK and glucose metabolism in skeletal muscle, based on results from lipid-depleted muscle cells [23].
At first glance, our findings in vitro appear difficult to reconcile with the glucose-lowering and insulin-sensitising effects of prolonged treatment with GW501516 in vivo [5, 25], because it is broadly accepted that increased fatty acid utilisation impairs glucose homeostasis via the glucose-fatty acid cycle. It is disputable whether the effect observed in isolated muscle is caused by insulin desensitisation, because the relative (%) reduction in glucose utilisation was similar in the absence and presence of insulin. However, there is no doubt that GW501516 considerably impaired the absolute rates of insulin-stimulated muscle glucose metabolism. The notion that such impairment will derange whole-body glucose homeostasis relates to studies in which increased fatty acid utilisation was triggered by the mass effect of elevated ambient lipid concentrations [20–22]. This clearly differs from the fuel switch induced by PPAR-δ, which occurs together with elevated glucose consumption and fatty acid synthesis in the liver [25] as well as with an increased capacity of the mitochondrial apparatus in skeletal muscle, which is therefore capable of oxidising larger amounts of lipid [5, 9, 11]. It is of note that these changes contrast with the fundamental derangement of insulin-resistant skeletal muscle, which is characterised by a distinctly reduced oxidative capacity and a pronounced preference for glucose over fatty acids as the oxidative substrate [26–28].
PPAR-δ-independent actions
Besides the PPAR-δ-mediated effects seen at low concentrations, we show that high concentrations of GW501516 affect cell respiration via direct, rapid, and hence PPAR-independent action on mitochondria. Direct effects on mitochondrial function have likewise been ascribed to agonists of PPAR-γ and PPAR-α, but whereas many of them seem to specifically impair the respiratory complex I [14, 17], GW501516 uncouples mitochondrial respiration from ATP synthesis independently of this enzyme complex. Inefficient ATP production due to uncoupling provides a plausible explanation of why parallel stimulation of both glucose and fatty acid oxidation at high concentrations of GW501516 is superimposed on the PPAR-δ-mediated shift from glucose to fatty acid oxidation at low concentrations. Parallel increases in the oxidation rates of both substrates clearly prevail after pre-exposure to 3 μmol/l GW501516 for 24 h. Higher concentrations of GW501516 were required to stimulate palmitate oxidation by 0.5 h of pre-exposure (10 μmol/l) or to induce uncoupling in isolated mitochondria, which were exposed to GW501516 for a few minutes only (30 μmol/l). Such a time-dependent increase in efficacy likewise characterises the mitochondrial effects of PPAR-γ agonists [14] and could be due to slow access and/or accumulation of these lipophilic compounds at their site of action (possibly the inner mitochondrial membrane).
Although our results do not necessarily imply that PPAR-independent uncoupling occurs under prolonged oral treatment in vivo, this possibility cannot be excluded, at least in animal studies, because plasma levels above 1.5 μmol/l GW501516 were reached in obese monkeys treated at 3 mg kg−1 day−1 [6], and the same or even higher doses have been used in rodents, showing, for example, an increase in oxygen consumption [4, 5]. Beyond conclusions that can be drawn for the specific agent GW501516, our findings support previous evidence that PPAR-independent effects on mitochondria should always be taken into account in the development of new ligands of all PPAR subtypes [14, 17].
Summary
In summary, we show that GW501516-induced activation of PPAR-δ switches mitochondrial substrate preference from carbohydrate to lipid, which contrasts with the reverse preference ascribed to insulin-resistant muscle and seems to relate to an increase in the oxidative capacity of mitochondria. Furthermore, GW501516 has a direct uncoupling effect on mitochondria, which could contribute to its metabolic actions in vivo.
Abbreviations
- AMPK:
-
AMP activated protein kinase
- EDL:
-
extensor digitorum longus
- FBS:
-
fetal bovine serum
- PPAR:
-
peroxisome proliferator-activated receptor
References
Evans RM, Barish GD, Wang YX (2004) PPARs and the complex journey to obesity. Nature Med 10:1–7
Ferré P (2004) The biology of peroxisome proliferator-activated receptors. Relationship with lipid metabolism and insulin sensitivity. Diabetes 53(Suppl 1):S43–S50
Luquet S, Gaudel C, Holst D et al (2005) Roles of PPAR delta in lipid absorption and metabolism: a new target for the treatment of type 2 diabetes. Biochim Biophys Acta 1740:313–317
Wang Y-X, Lee C-H, Tiep S et al (2003) Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell 113:159–170
Tanaka T, Yamamoto J, Iwasaki S et al (2003) Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci U S A 100:15924–15929
Oliver WR, Shenk JL, Snaith MR et al (2001) A selective peroxisome proliferator-activated receptor δ agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A 98:5306–5311
Leibowitz MD, Fiévet C, Hennuyer N et al (2000) Activation of PPARδ alters lipid metabolism in db/db mice. FEBS Lett 473:333–336
Dressel U, Allen TL, Pippal JB, Rohde PR, Lau P, Muscat GEO (2003) The peroxisome proliferator-activated receptor β/δ agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol Endocrinol 17:2477–2493
Wang Y-X, Zhang C-L, Yu RT et al (2004) Regulation of muscle fiber type and running endurance by PPARδ. PLoS Biol 2:e294
Muoio DM, MacLean PS, Lang DB et al (2002) Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) α knock-out mice. Evidence for compensatory regulation by PPARδ. J Biol Chem 277:26089–26097
Luquet S, Lopez-Soriano J, Holst D et al (2003) Peroxisome proliferator-activated receptor δ controls muscle development and oxidative capability. FASEB J 17:2299–2301
Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN (2004) Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-δ accelerates intestinal adenoma growth. Nat Med 10:245–247
Crettaz M, Prentki M, Zanietti D, Jeanrenaud B (1980) Insulin resistance in soleus muscle from obese Zucker rats. Involvement of several defective sites. Biochem J 186:525–534
Brunmair B, Staniek K, Gras F et al (2004) Thiazolidinediones, like metformin, inhibit respiratory complex I. A common mechanism contributing to their antidiabetic actions? Diabetes 53:1052–1059
Brunmair B, Gras F, Neschen S et al (2001) Direct thiazolidinedione action on isolated rat skeletal muscle fuel handling is independent of peroxisome proliferator-activated receptor-γ-mediated changes in gene expression. Diabetes 50:2309–2315
Lowry OH, Passoneau JV (1972) A flexible system of enzymatic analysis. Academic, New York
Brunmair B, Lest A, Staniek K et al (2004) Fenofibrate impairs rat mitochondrial function by inhibition of respiratory complex I. J Pharmacol Exp Ther 311:109–114
Gilde AJ, van der Lee KAJM, Willemsen PHM et al (2003) Peroxisome proliferator-activated receptor (PPAR) α and PPARδ/β, but not PPARγ, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res 92:518–524
Forman BM, Chen J, Evans RM (1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and γ. Proc Natl Acad Sci U S A 94:4312–4317
Randle PJ, Garland PB, Hales CN, Newsholme EA (1963) The glucose-fatty acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 281:785–789
Roden M, Price TB, Perseghin G et al (1996) Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 97:2859–2865
Krebs M, Krssak M, Nowotny P et al (2001) Free fatty acids inhibit the glucose-stimulated increase of intramuscular glucose-6-phosphate concentration in humans. J Clin Endocrinol Metab 86:2153–2160
Krämer DK, Al-Khalili L, Perrini S et al (2005) Direct activation of glucose transport in primary human myotubes after activation of peroxisome proliferator-activated receptor δ. Diabetes 54:1157–1163
Son C, Hosoda K, Matsudo J et al (2001) Up-regulation of uncoupling protein 3 gene expression by fatty acids and agonists for PPARs in L6 myotubes. Endocrinology 142:4189–4194
Lee C-H, Olson P, Hevener A et al (2006) PPARδ regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci USA 103:3444–3449
Kelley DA, Simoneau J-A (1994) Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J Clin Invest 94:2349–2356
Kelley DA, Goodpaster B, Wing RR, Simoneau J-A (1999) Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol 277:E1130–E1141
Kelley DE, Mandarino LJ (2000) Fuel selection in human skeletal muscle in insulin resistance. A reexamination. Diabetes 49:677–683
Acknowledgement
We thank Hoffmann-LaRoche Research and Development (Basel, Switzerland) for generously providing GW501516, and the staff at the Biomedical Research Centre, Medical University of Vienna, for taking care of the rats. This work was supported by the Austrian Science Fund (grant no. P16352-B08).
Duality of interest.The authors declare that they have no duality of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Brunmair, B., Staniek, K., Dörig, J. et al. Activation of PPAR-δ in isolated rat skeletal muscle switches fuel preference from glucose to fatty acids. Diabetologia 49, 2713–2722 (2006). https://doi.org/10.1007/s00125-006-0357-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0357-6