
Abstract
RBM20 is one of the genes predisposing to dilated cardiomyopathy (DCM). Variants in the RS domain have been reported in many DCM patients, but the pathogenicity of variants within the RNA-recognition motif remains unknown. Two human patients with the I536T-RBM20 variant without an apparent DCM phenotype were identified in sudden death cohorts. A splicing reporter assay was performed, and an I538T knock-in mouse model (Rbm20I538T) was generated to determine the significance of this variant. The reporter assay demonstrated that the human I536T variant affected the TTN splicing pattern compared to wild-type. In the mouse experiments, Rbm20I538T mice showed different splicing patterns in Ttn, Ldb3, Camk2d, and Ryr2. The expressions of Casq1, Mybpc2, and Myot were upregulated in Rbm20I538T mice, but Rbm20I538T mice showed neither DCM nor cardiac dysfunction on histopathological examination and ultrasound echocardiography. The I536T-RBM20 (I538T-Rbm20) variant changes gene splicing and affects gene expression, but the splicing and expression changes in Ttn and Ca handling genes such as Casq1, Camk2d, and Ryr2 do not cause DCM morphology in the mouse model.
Key messages
• Two human patients with the I536T-RBM20 variant without a DCM phenotype were identified.
• A splicing reporter assay demonstrated that the variant affected the TTN splicing.
• Rbm20I538T mice showed neither DCM nor cardiac dysfunction.
• Rbm20I538T mice showed different splicing patterns and the gene expressions.
Similar content being viewed by others


Introduction
Sudden unexpected death in a young adult is a rare event, but elucidating its mechanisms is one of the most important issues in medical science. Recently, genetic analysis has been improved and adopted in the field of postmortem examination, and there are many reports and studies in which postmortem genetic analysis uncovered the mechanism of autopsy-negative sudden death [1,2,3,4]. Most studies used target sequencing and exome sequencing with next-generation sequencing. However, problems arise when comprehensive exome sequencing detects many genetic variants, and the interpretation becomes difficult [5]. The mechanism at the molecular biological level is important.
RBM20, encoding RNA Binding Motif Protein-20 (RBM20), is the regulator of heart-specific pre-mRNA splicing [6]. Mutations in this gene were identified in human-dilated cardiomyopathy (DCM, MIM613172) patients [7]. Since these first patients with RBM20 variants [7], several dozen variants have been reported so far [8], most of which were associated with DCM phenotypes.
One of the target genes of RBM20 is TTN [6, 9,10,11]. The protein product of the TTN gene, titin, composes part of the sarcomere and also acts as a molecular spring. It has the largest number of exons in humans and has various types of splice variants. The isoforms of cardiac types include the shorter N2B and the longer N2BA isoforms, and that of the skeletal muscle types includes the N2A isoform. Abnormal splicing of TTN caused by RBM20 variants is considered to contribute to DCM phenotypes. In the mouse model with RBM20 pathogenic variants, an abnormal giant N2BA isoform, the N2BA-G isoform, is predominant. It results from inclusion of apparently all the alternatively spliced exons of Ig and PEVK (proline, glutamate, valine, lysine) regions, all of which are not included in the normal cardiac isoforms. This giant molecular spring provides reduced passive stiffness to the cardiomyocyte, contributing to DCM [12].
The mutation hotspot region of RBM20 is within the RS domain [8, 13], among which P633L, R634Q, R634W, R634L, S635A, R636S, R636C, R636H, S637G, and P638L are within the RSRSP stretch. In contrast, few RBM20 disease variants have been found within the RNA-recognition motif (RRM) domain. Of the RBM20-related DCMs, the variants in RS-domain show familial DCM, whereas a single example of a V535I variant was found in a sporadic DCM patient [14]. The G583C variant was also reported in a hypertrophic cardiomyopathy (HCM) patient [15]. Recently, we also reported a sudden death patient with a heterozygous I536T variant, situated in the strictly conserved RRM region [16]. This sudden death patient with the I536T variant did not show DCM morphologically, and thus, the effect of I536T-RBM20 on the cardiac function is controversial.
In this study, molecular biological analysis was performed by using a splicing reporter assay, and then an I538T knock-in (KI) mouse model (Rbm20I538T) was generated to determine the relevance of this variant.
Materials and methods
Editorial policies and ethical considerations
All research was performed in accordance with the guidelines for human genome analysis in Japan, and the study protocol was approved by the Ethics Committee of Hyogo Medical University (0358) and Gunma University Graduate School of Medicine (2017–015). Written, informed consent for genetic analysis was obtained from the relatives. All animal experiments were performed with the approval of the Hyogo Medical University Institutional Animal Care and Use Committee (18–078) and the ARRIVE guideline.
Identification of RBM20 variants in human sudden death patients
Next-generation sequencing was used to screen sudden death patients for variants in exons and exon–intron boundaries of RBM20. The variants detected were confirmed by Sanger sequencing, as described elsewhere.
Plasmid construction
The TTNE50-E51E218-E219-EGFP/mCherry minigene reporter plasmid was constructed as previously described [9]. Expression plasmids for human RBM20 protein pRP[Exp]-Puro-CAG > hRBM20[NM_001134363.3] were purchased from VectorBuilder Inc (Chicago, IL). Using overlapping PCR mutagenesis, the mutations of c.T1607C were inserted into the wild-type cDNA in constructs of pRP-hRBM20I536T. For all constructs, sequencing was performed over the entire region of the amplified sequences. Plasmid DNA was purified using a HiSpeed® Plasmid Maxi Kit (Qiagen, Venlo, Netherland).
Cell culture and transfection
HeLa cells were cultured in DMEM Medium (FUJIFILM Wako Pure Chemical, Osaka, Japan) supplemented with 10% fetal bovine serum at 37 °C with 5% CO2. The TTN reporter minigene and the expression vectors were transiently co-transfected in a 1:4 mixture into Hela cells using Lipofectamine (Thermo Fisher Scientific, Waltham, MA) in accordance with the manufacturer’s protocol. Fluorescence images of fluorescent proteins were acquired 24 h after transfection using EVOS (Thermo Fisher Scientific), and then the cells were harvested for total RNA preparation and RT-PCR analysis.
Total RNA extraction and RT-PCR of the reporter assay
Total RNAs from HeLa cells involved in transient transfection experiments were extracted using the RNeasy mini kit (Qiagen). Complementary DNAs (cDNAs) were prepared with the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific) using 1 µg of RNA with a random primer, in accordance with the manufacturer’s protocol. Semi-quantitative PCRs were performed using PrimeStarGXL (Takara Bio Inc., Shiga, Japan), and the PCR products were analyzed using the Bioanalyzer 2100 Expert with DNA7500 Kit (Agilent Technologies Inc., Santa Clara, CA). Primers and conditions of RT-PCR were described previously [9].
Animal procedures
Mice were kept in a temperature-controlled facility under specific-pathogen-free conditions with ad libitum access to food and water in a 12-h light/dark cycle. C57BL/6 J mice were purchased from CLEA Japan (Tokyo, Japan).
Preparation of Cas9/gRNA complex and ssDNA
The gRNA was designed using CRISPRdirect (https://crispr.dbcls.jp) to target exon 6 of Rbm20. The gRNA sequence was as follows: Rbm20 gRNA 5′-GGAGAATGACGTCATTAACC-3′. The corresponding crRNA (5′-GGAGAAUGACGUCAUUAACCGUUUUAGAGCUAUGCU-3′), tracrRNA (#1,072,533), and Cas9 nuclease were purchased from Integrated DNA Technologies (IDT, Coralville, IA). The crRNA and tracrRNA were annealed and complexed with Cas9 nuclease according to the manufacturer’s instructions.
Single-strand DNA (ssDNA) with c.T1613C mutation was ordered as PAGE Ultramer from IDT. The ssDNA sequence was as follows: Rbm20 ssDNA 5′-GCACATCTGCAATCTCCCGGAGGGCAGCTGCACGGAGAATGACGTCACTAACCTGGGGCTGCCCTTTGGCAAGGTCACTAATTACATCCTCATGAAGTCA-3′.
Electroporation
Male and female C57BL/6 mice (CLEA Japan) were used as sperm and oocyte donors. Female mice were superovulated, and then zygotes were prepared by in vitro fertilization. They were cultured in KSOM medium (ARK Resource, Kumamoto, Japan) before and after electroporation. Electroporation was performed using a 1-mm gap electrode (CUY501P1-1.5) and a super electroporator NEPA21 (Nepa Gene, Chiba, Japan). Zygotes were washed with Opti-MEM (Thermo Fisher Scientific) and then placed in the electrode gap filled with 5 µl of Opti-MEM solution containing 200 ng/µL Cas9 protein, 6 pmol/µL gRNA (crRNA/tracrRNA complex), and 10 pmol/µL ssDNA. They were cultured in KSOM medium overnight, and surviving 2-cell-stage embryos were transferred to the oviducts of pseudopregnant Jcl:ICR female mice (CLEA Japan).
Genomic DNA extraction, genotyping, and sequencing
Genomic DNA was extracted from F0, F1, and F2 mice. PCR was performed using Taq DNA polymerase (Greiner Bio-One, Kremsmünster, Austria). The primer sequences are shown in Supplementary Material, Table S1. The PCR products were purified using the Gel/PCR Extraction Kit (NIPPON Genetics, Tokyo, Japan). The purified PCR products were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) through an Applied Biosystems 3500xL Genetic Analyzer (Thermo Fisher Scientific), or restriction enzyme digestion was performed by RspRS II enzyme (Takara Bio Inc.), according to the manufacturer’s instructions. In some cases, PCR products were cloned into TA vector (Promega, Madison, WI) before sequencing.
Total RNA extraction and RNA sequencing
After euthanasia, total RNAs from mouse hearts at the ages of 1, 12, and 36 weeks were extracted using the RNeasy Plus Universal Mini Kit (Qiagen). Nine representative libraries were generated for the nine groups (wild-type, heterozygote, and homozygote of 1 week, 12 weeks, and 36 weeks) by RNA-seq with SMART-seq v4 Ultra Low Input RNA kit for sequencing (Takara Bio Inc.) and Nextera XT DNA Library Prep Kit (Illumina, San Diego, CA) and sequenced on a NovaSeq 6000 instrument (Illumina).
Bioinformatic analysis
Splicing analysis with Percentage Spliced In (PSI) was performed as described previously [16]. Differential gene expression analysis was performed using StrandNGS (Strand Life Sciences Pvt. Ltd., Bangalore, India). The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE201018.
Splicing analysis
Complementary DNA was synthesized from total RNA (1 µg) using a Prime-Script RT PCR Kit (Takara Bio Inc.), according to the manufacturer’s instructions. The confirmation of the quantification of the splicing variant was performed as previously described [16]. In detail, total RNA was reverse transcribed to cDNA, and PCR amplification was performed using PrimeSTAR Max Premix (2) (Takara Bio Inc.), with the primers shown in Supplementary Material, Table S2. Each splicing variant of the PCR product was shown using an Agilent 2100 Bioanalyzer performed with a DNA 1000 LabChip Kit and High sensitivity LabChip Kit (Agilent Technologies Inc.). For semi-quantification, the proportions of splicing variant were calculated using the following formula: proportion = amount of the one product/ (amount of the one product + amount of the other product).
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed using TB Green® Premix Ex Taq™ II (Takara Bio Inc.) and a Thermal Cycler Dice Real Time System (Takara Bio Inc.). Sequences of the PCR primers used are presented in Supplementary Material, Table S3. Expression levels were normalized with Ttn exon50 or Gapdh. Statistical significance was assessed by Prism9.
Histopathology
For the histological analyses, heart tissue at the age of 12 weeks was fixed in paraformaldehyde, 4% in phosphate-buffered saline, embedded in paraffin, sectioned, and subjected to hematoxylin–eosin staining and Masson’s trichrome staining. Stained sections were photographed using Virtual Slide System VS120 (Olympus Scientific Solutions, Tokyo, Japan).
Cardiovascular phenotyping
Ultrasound echocardiography (UCG) was performed in a blinded manner. In detail, mice at the age of 12 weeks were anesthetized with isoflurane and placed on a heated platform. Cardiac functions, such as left ventricular fractional shortening (LVFS), end-diastolic dimension (LVDd), and diastolic wall thickness of the LV posterior wall (LVPWth), were assessed using a high-frequency ultrasound system (Vevo 2100 with MS400, FUJIFILM VisualSonics, Inc., Toronto, Canada). A two-dimensional parasternal short-axis view was obtained at the levels of the papillary muscles, and M-mode tracings were recorded.
Results
Human cases of RBM20 I536T presented sudden death without apparent DCM
Two Japanese sudden death patients who possessed the I536T-RBM20 variant were identified. The first patient was reported previously [16]. The second patient was a male in his 20 s, who had no history of arrhythmia (Supplementary Material, Fig. S1) or collapse. He was found dead in bed during sleep. He had no family history of sudden death or arrhythmia. His weight was 58 kg, and his height was 167 cm. The heart weighed 320 g (Fig. 1A). No cardiac dilatation was observed (Fig. 1B). The coronary arteries showed no stenoses. The left and right lungs weighed 610 g and 640 g, respectively, and the edematous brain weighed 1670 g. Microscopic examination showed disarray and diffuse mild fibrosis in the heart (Fig. 1C).

Human sudden death case. A, B Macroscopic examination of heart A and computed tomography image B. No cardiac dilatation is observed DCM. C Microscopic examination shows disarray and diffuse mild fibrosis in the heart. D Postmortem genetic analyses show the I536T-RBM20 variant. It is conserved among the species
Postmortem genetic analyses showed that both patients had the I536T-RBM20 variant (Fig. 1D). The familial analyses were not allowed. The residue was conserved among the species and was located in the RRM region. In silico prediction programs suggested that this variant is likely pathogenic: probably damaging by PolyPhen-2 and Deleterious by SIFT.
Human RBM20 I536T mutation affects TTN splicing in the splicing reporter assay
To evaluate the effect of the RBM20 I536T mutation on the splicing of the TTN gene, in vitro assays were performed using a dichromatic Ttn alternative splicing reporter minigene. Human RBM20 wild-type, as well as I536T mutant, vectors were co-transfected with the reporter minigene vector into Hela cells. Green fluorescent protein (EGFP) is expressed from the product containing the fused exon E51E218, and red fluorescent protein (mCherry) is translated from the E51E218-skipped mRNA, corresponding to the splicing patterns of the N2A and N2B types, respectively (Fig. 2A).

Dichromatic fluorescence alternative splicing reporter for Ttn to monitor the RBM20 activity in splicing regulation. A Schematic representation of the Ttn reporter minigene TtnE50-E51E218-E219-EGFP/mCherry (top) and mRNAs derived from it (bottom). Two genomic fragments Ttn E50-E51 and E218-E219 are inserted between human histone H2B cDNA and the EGFP/mCherry cassette. Expression of E51E218EGFP and ΔE51E218-mCherry indicates inclusion and skipping of a chimeric exon E51E218, respectively. B Microphotographs of HeLa cells co-transfected with the fluorescence Ttn splicing reporter minigene and an empty vector or an expression vector for the wild-type or mutant RBM20 protein. Fluorescence of EGFP and mCherry is pseudo-colored in green and magenta, respectively. The experiment was performed twice. C RT-PCR analysis of the TTN splicing reporter minigene co-expressed with an empty vector or an expression vector for the wild-type or mutant RBM20 protein in HeLa cells. Representative gel-like presentations are shown. The ratio of N2A/N2B is shown. The N2A type is not detected with the wild-type RBM20, whereas it is still detected with the I536T mutant vector
In Hela cells co-transfected with an empty vector, fluorescence of both N2A-type EGFP and N2B-type mCherry was detected (Fig. 2B). Co-expression of human wild-type RBM20 promoted the expression of N2B-type mCherry and suppressed expression of N2A-type EGFP (Fig. 2B, middle panel). On the other hand, co-expression of the human I536T mutant vector less efficiently promoted expression of N2B-type mCherry, and expression of N2A-type EGFP still remained (Fig. 2B, right panel). In addition, the splicing pattern of mRNAs derived from the minigene was examined by using semi-quantitative RT-PCR. The remaining expression of N2A type by transfection of the I536T mutant vector was confirmed by electrophoresis, and N2A/N2B ratio was increased in the I536T mutant vector (Fig. 2C). These results suggested that human RBM20 I536T affects the splicing of TTN.
Generation of the KI mice
Using the CRISPR/Cas9 system, KI mice harboring the c.T1613C (p.I538T) point mutation in Rbm20, which corresponded to human c.T1607C (p.I536T), were generated (Supplementary Material, Fig. S2 and Fig. S3). Through electroporation of the Cas9/gRNA complex and donor ssDNA into zygotes, 36 founder (F0) mice were born. Of them, 14 mice (39%) had the intended point mutation. These F0 mice were crossed with C57BL/6 J mice (CLEA Japan), and heterozygous F1 mice, which were confirmed by Sanger’s sequencing, were obtained.
The KI mice were born according to Mendel’s law and were all healthy, fertile, and indistinguishable from their wild-type littermates.
Rbm20 KI mice showed abnormal splicing
To exclude the effect of altered Rbm20 expression on splicing of other genes, qRT-PCR was performed, and there were no differences in Rbm20 expression among the three genotypes (Fig. 3A).

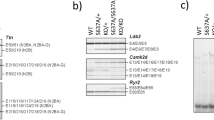
Rbm20.I538T knock-in mice show abnormal splicing in the heart (RNA-seq analysis). A Rbm20 RNA levels normalized to Gapdh and wild-type levels are not different on qRT-PCR in the heterozygote and the homozygote. n = 3 each. B–E The average PSI of 3 mice per genotype is shown (wild-type in blue, heterozygote in red, homozygote in green), and the resulting ΔPSIs (wild-type vs. homozygotes) show abnormal exon inclusion or exclusion in Ttn B, Ldb3 C, Camk2d D, and Ryr2 E
To determine whether the RBM20 variant affects the splicing of the target genes such as Ttn, RNA-seq was performed on heart samples from three genetic types of each age. Splicing analysis with PSI showed a higher percentage for the Ttn exons encoding I band region (ΔPSI of approximately 60% between wild-type and homozygous type), and various abnormal splicing patterns in Ldb3, Camk2d, and Ryr2 were also detected (Fig. 3B–E), which were also detected in other KI mice [17].
To validate this abnormal splicing, RT-PCR was performed and confirmed the abnormal splicing patterns of these genes. In addition, qRT-PCR showed that inclusion of Ttn exons 51, 52, and 53 was increased in the homozygous type compared to the wild-type (Fig. 4A, B).


Rbm20I538T knock-in mice have abnormal splicing in the heart (RT-PCR validation). A Schematic representation of the Ttn isoform and the re-indication of the RNA-seq data are shown. The bands of RT-PCR analysis are detected in the homozygote (left, exon50-53) and in the wild-type and heterozygote (right, exon50-219). B Relative expression of Ttn exons 51, 52, and 53 compared to exon50 shows that these exons are more included in the homozygote than the wild-type and the heterozygote. n = 6 each. C Schematic representation of the Ldb3 isoforms. The RT-PCR analysis shows that exon4 (cardiac isoform, 604 bp) is included in all genotypes, whereas exons 5, 6, and 7 (skeletal isoform, 487 bp) are included predominantly in the homozygote, which was hardly detected in the wild-type or the heterozygote. The inclusion proportion is significantly different among the genotypes. n = 6 each. The RT-PCR analyses of exon11 inclusion/exclusion are shown, and there is no significant difference among the genotypes. n = 6 each. D The RT-PCR analysis of Camk2d mRNAs shows isoform differences among the genotypes. The graph shows the proportion of each isoform. E The RT-PCR analysis of Ryr2 mRNAs shows abnormal 24-bp exon inclusion in the homozygote. There is significant difference among the genotypes. n = 3 each. Error bars indicate 95% confidence interval, Tukey’s multiple comparison test
Exon 4 of Ldb3 is included in the cardiac isoform, whereas exons 5, 6, and 7 are included instead in the skeletal isoform. PSI analysis and RT-PCR showed an abnormal splicing pattern in the homozygotes, i.e., inclusion of exons 5, 6, and 7, in the cardiac tissue (Fig. 4C). To confirm the isoforms with exon 11 inclusion/exclusion, RT-PCR was performed from exon 8 to exon 13 of Ldb3 and showed that the inclusion/exclusion ratio of exon 11 of Ldb3 was not different among the three genotype groups (Fig. 4C).
Camk2d has several types of isoforms [18, 19]. PSI analysis and RT-PCR showed that the longer isoform including exons 13, 15, 16, and 17 (1A) increased in the homozygotes, whereas the shorter one including exons 13 and 17 (2C) was predominant in the wild-type (Fig. 4D).
The homozygous type included an additional 24-bp exon in the RyR2 gene, which was confirmed by RT-PCR (Fig. 4E).
Three mRNAs were upregulated in the homozygous Rbm20 KI mice
To compare the gene expression profiles in Rbm20 KI mice, RNA-seq from the heart was performed at 1, 12, and 36 weeks of age. A total of 1079 differentially expressed genes (DEGs) between the WT and Rbm20 KI mice (270 upregulated and 809 downregulated genes) were identified (Fig. 5A). Of them, 5 genes were upregulated at every stage of age, including Casq1, Mybpc2, and Myot, which were related to heart functions. These three upregulated genes were then examined, and qRT-PCR showed that the expressions of Casq1, Mybpc2, and Myot were significantly increased in the homozygotes (Fig. 5B). Nmrk2, which was highly expressed in DCM [20], was also upregulated in the RNA-seq analysis, but qRT-PCR analysis showed no significant difference (Fig. 5C).

Gene expression profile analysis with RNA-seq data. A Venn diagram analysis of differentially expressed genes at 3 weeks (left, upregulated genes; right, downregulated genes). B qRT-PCR analysis of upregulated genes at the age of 12 weeks. n = 6 each. C qRT-PCR analysis of Nmrk2 genes at the age of 12 weeks. n = 6 each. Error bars indicate 95% confidence interval, Tukey’s multiple comparison test
Rbm20 KI mice did not show sudden death or a cardiac phenotype
None of the Rbm20 KI heterozygous or homozygous mice experienced sudden death until the age of 36 weeks (n = 6), which is different from the human sudden death cases with the same RBM20 variant. Heart weight was not different among genotypes (Fig. 6A). Pathological examination showed no cardiac dilation and subendocardial fibrosis (Fig. 6B). The expressions of Col1a2, Col3a1, and Mmp2 mRNAs were not increased in the Rbm20 KI mice (Fig. 6C), consistent with the lack of fibrosis.

Rbm20I538T knock-in mice do not show cardiac dysfunction. A Heart/body weight ratio at the age of 12 weeks. n = 6 each. B Representative images of H&E staining and Masson’s trichrome staining at the age of 12 weeks. Scale bars, 4 mm. C qRT-PCR analysis of genes related to cardiac fibrosis at the age of 12 weeks. n = 6 each. D Measured values of LVFS, LVDd, and LVPWth by UCG at the age of 12 weeks. n = 6 each. E Representative images of UCG at the age of 12 weeks. F Gene expression of Nppa and Nppb in the heart assessed by qRT-PCR at the age of 12 weeks. n = 6 each. Error bars indicate 95% confidence interval. Tukey’s multiple comparison test
To assess the cardiac phenotypes of the Rbm20 mutant mice, UCG was performed. The UCG data showed no significant difference in LVFS, LVDd, and LVPWth (Fig. 6D, E). Indeed, LVFS of some of the homozygous mice seemed to be decreased, as shown in Fig. 6D, though there were no significant differences because of the small sample size. However, the relative expressions of Nppa and Nppb, which are biomarkers of heart failure, were not upregulated (Fig. 6F).
Discussion
In the present study, two human cases of sudden death with I536T-RBM20 were presented, and, using in vitro and in vivo analyses, it was demonstrated that this variant affected alternative splicing and gene expression without an apparent cardiac phenotype.
There are three main issues to be addressed. First, patient 2 presented with sudden death, and genetic analysis showed I536T-RBM20, but splicing analysis was not performed, and the splicing effect was unknown. Second, patient 1, described elsewhere [16], had an asymptomatic genetic background of myotonic dystrophy type 1 (DM1) with expanded CTG repeats in the DMPK gene, in addition to I536T-RBM20. Because both genetic predispositions affect exon splicing [21], it was not possible to determine with any certainty whether the abnormal splicing of patient 1 was caused by I536T-RBM20 alone [16]. Third, we have no doubt that RBM20 is one of the important causative genes of DCM [7], but the I536T-RBM20 variant has never been reported, and the exact influence of this variant on DCM is still unknown. One goal of this study was to uncover the splicing effect in humans by using a reporter assay, and another major goal was to perform an analysis using a KI mouse model.
First, the effect of the I536T variant on human TTN splicing was surveyed. In patient 1, it was possible to obtain a frozen sample of heart tissue and perform RNA-seq analysis to calculate PSI. The splicing pattern of TTN in patient 1 was not the same as that in the control sample [16]. As for patient 2, because it was not possible to obtain a frozen sample, a reporter assay was performed as an alternative. The reporter assay showed abnormal Ttn splicing, which was consistent with the cardiac sample from patient 1. These results of the I536T variant were similar to other reports that abnormal TTN splicing was detected in patients with cardiac phenotypes who have mutations in RBM20 [11, 22, 23], indicating that the I536T variant caused aberrant alternative splicing even in non-DCM patients.
Next, a KI mouse model of I538T-Rbm20 was generated. There have been several types of animal models of RBM20 cardiomyopathy [6, 17, 24,25,26,27,28,29,30]. Knock-out (KO)-model mice and rats showed abnormal splicing with only mild DCM phenotypes [6, 17, 24, 26, 27, 29]. KI mice and pigs with mutations in the RS region, such as R636S, S637A, and S639G, well mimicked human DCM phenotypes [17, 28, 30]. On the other hand, an RRM domain-deletion mouse model also showed the abnormal splicing in Ttn, Camk2d, and Ldb3, but did not show the DCM phenotype, such as LV chamber dilatation and systolic dysfunction [25].
In the present study, Rbm20I538T/I538T mice showed abnormal splicing in Ttn, Ldb3, Camk2d, and Ryr2. The splicing in Camk2d showed change to the longer isoform of Camk2d-1A, and Ryr2 included the additional 24-bp exon. These two genes are related to Ca2+ handling [11, 26, 27], which induces cardiac arrhythmias. The shift in CamkIIδ isoforms largely underlies the Ca2+ handling defects in another KO mouse model of splicing factors [31]. RNA-seq analysis showed that the expressions of Casq1, Mybpc2, Myot, and Nmrk2 increased during the animals’ lifetime (1 week to 36 weeks of age). The clinical significance of the increase in these genes remains unknown, but Casq1, Mybpc2, and Myot were also increased in the other studies [17], and upregulation of these genes was validated. CASQ is one of the calcium-binding proteins, and both isoforms, CASQ1 and CASQ2, interact with the ryanodine receptors calcium release channels [32]. Human patients with RBM20 variants are susceptible to arrhythmias such as ventricular tachycardia, leading to cardiac transplantation, implantation of an implantable cardioverter-defibrillator, or sudden death [7, 14, 27, 33,34,35,36]. Ventricular arrhythmias caused by RBM20-associated abnormal splicing of ion channels genes such as RYR2 were also reported [37]. Thus, it has not yet been precluded that Rbm20I538T/I538T mice have susceptibility to arrhythmia.
On the contrary, Rbm20I538T/I538T mice did not show DCM, although RBM20 cardiomyopathy often shows DCM phenotypes. Nmrk2, which was highly expressed in DCM [20], showed no significant difference on qRT-PCR analysis, though it was upregulated in RNA-seq analysis. Other biomarkers of heart failure, Nppa and Nppb, were also not upregulated. Considering the results of biophysical and morphological examinations, heart failure did not happen in Rbm20I538T/I538T mice. In this respect, the human I536T patient and I538T mice are not typical.
In addition, Rbm20I538T/I538T mice showed abnormal Ldb3 splicing of exons 3, 4, 5, and 6, but they did not show a significant difference in the exon 11 inclusion/exclusion ratio. Abnormal splicing of exons 3 to 6 was reported in RBM20 cardiomyopathy [6], and that of exon 11 was reported in myotonic dystrophy [38]. Patient 1 showed abnormal splicing in exon 11 [16]. The present experiment explained that this abnormal exon 11 splicing was not caused by the RBM20 variant but was possibly caused by myotonic dystrophy.
This study has some future implications and limitations. This variant has not been reported, but the fact that these two patients were encountered at two independent institutes, and they were not consanguineous, means that there are possibly more sudden death cases with this variant. Both patients had a heterozygous I536T variant, which was thought to have been inherited from one of the parents without a familial history of cardiomyopathy or sudden death, though familial genetic analysis was not permitted. Thus, just having this variant does not seem to cause sudden death, and an unknown factor seemed to be a trigger. Rbm20I538T/I538T mice were generated, and it was demonstrated that Rbm20I538T/I538T mice mimicked human I536T-RBM20 heterozygous patients in morphological phenotype and abnormal splicing, but they differed in that they did not suffer sudden death. This mouse model will have a great advantage in further studies of elucidating mechanisms of sudden death. Patient 1 had another genetic background, myotonic dystrophy. We would like to generate a double-transgenic mouse with Rbm20 and Dmpk in the future. Investigating the genetic background, as well as uncovering the trigger of sudden death, is an important issue to diagnose the cause of death from congenital diseases. On the other hand, molecular biological analyses and morphological investigations were performed, but electrocardiograms were not analyzed. Further examination would be necessary.
In conclusion, two sudden death cases of human I536T-RBM20 variant were presented. Splicing analysis of patient 2 was not performed directly, but the reporter assay showed an abnormal Ttn splicing. The mouse model showed abnormal splicing and different gene expressions without sudden death or cardiac failure. These splicing and expression changes in Ttn and Ca handling genes do not cause DCM morphology.
Data availability
The data sets used and analyzed during the current study are available from the corresponding author upon request. The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE201018.
References
Bagnall RD, Das KJ, Duflou J, Semsarian C (2014) Exome analysis–based molecular autopsy in cases of sudden unexplained death in the young. Heart Rhythm 11:655–662. https://doi.org/10.1016/j.hrthm.2014.01.017
Narula N, Tester DJ, Paulmichl A et al (2015) Post-mortem whole exome sequencing with gene-specific analysis for autopsy-negative sudden unexplained death in the young: a case series. Pediatr Cardiol 36:768–778. https://doi.org/10.1007/s00246-014-1082-4
Hata Y, Kinoshita K, Mizumaki K et al (2016) Postmortem genetic analysis of sudden unexplained death syndrome under 50 years of age: a next-generation sequencing study. Heart Rhythm 13:1544–1551. https://doi.org/10.1016/j.hrthm.2016.03.038
Oshima Y, Yamamoto T, Ishikawa T et al (2017) Postmortem genetic analysis of sudden unexpected death in infancy: neonatal genetic screening may enable the prevention of sudden infant death. J Hum Genet 62:989–995. https://doi.org/10.1038/jhg.2017.79
Shingu K, Murase T, Yamamoto T et al (2021) Accurate interpretation of genetic variants in sudden unexpected death in infancy by trio-targeted gene-sequencing panel analysis. Sci Rep 11:21532. https://doi.org/10.1038/s41598-021-00962-8
Guo W, Schafer S, Greaser ML et al (2012) RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 18:766–773. https://doi.org/10.1038/nm.2693
Brauch KM, Karst ML, Herron KJ et al (2009) Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol 54:930–941. https://doi.org/10.1016/j.jacc.2009.05.038
Jordà P, Toro R, Diez C et al (2021) Malignant arrhythmogenic role associated with RBM20: a comprehensive interpretation focused on a personalized approach. J Pers Med 11:130. https://doi.org/10.3390/jpm11020130
Murayama R, Kimura-Asami M, Togo-Ohno M et al (2018) Phosphorylation of the RSRSP stretch is critical for splicing regulation by RNA-Binding Motif Protein 20 (RBM20) through nuclear localization. Sci Rep 8:8970. https://doi.org/10.1038/s41598-018-26624-w
Li S, Guo W, Dewey CN, Greaser ML (2013) Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res 41:2659–2672. https://doi.org/10.1093/nar/gks1362
Maatz H, Jens M, Liss M et al (2014) RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest 124:3419–3430. https://doi.org/10.1172/JCI74523
Guo W, Sun M (2018) RBM20, a potential target for treatment of cardiomyopathy via titin isoform switching. Biophys Rev 10:15–25. https://doi.org/10.1007/s12551-017-0267-5
Watanabe T, Kimura A, Kuroyanagi H (2018) Alternative splicing regulator RBM20 and cardiomyopathy. Front Mol Biosci 5:105. https://doi.org/10.3389/fmolb.2018.00105
Li D, Morales A, Gonzalez-Quintana J et al (2010) Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci 3:90–97. https://doi.org/10.1111/j.1752-8062.2010.00198.x
Dai J, Li Z, Huang W et al (2021) RBM20 is a candidate gene for hypertrophic cardiomyopathy. Can J Cardiol 37:1751–1759. https://doi.org/10.1016/j.cjca.2021.07.014
Yamamoto T, Miura A, Itoh K et al (2019) RNA sequencing reveals abnormal LDB3 splicing in sudden cardiac death. Forensic Sci Int 302:109906. https://doi.org/10.1016/j.forsciint.2019.109906
Ihara K, Sasano T, Hiraoka Y et al (2020) A missense mutation in the RSRSP stretch of Rbm20 causes dilated cardiomyopathy and atrial fibrillation in mice. Sci Rep 10:17894. https://doi.org/10.1038/s41598-020-74800-8
Gray CBB, Heller Brown J (2014) CaMKIIdelta subtypes: localization and function. Front Pharmacol. https://doi.org/10.3389/fphar.2014.00015
Duran J, Nickel L, Estrada M et al (2021) CaMKIIδ splice variants in the healthy and diseased heart. Front Cell Dev Biol 9:644630. https://doi.org/10.3389/fcell.2021.644630
Tannous C, Deloux R, Karoui A et al (2021) NMRK2 gene is upregulated in dilated cardiomyopathy and required for cardiac function and NAD levels during aging. Int J Mol Sci 22:3534. https://doi.org/10.3390/ijms22073534
López-Martínez A, Soblechero-Martín P, de-la-Puente-Ovejero L, et al (2020) An overview of alternative splicing defects implicated in myotonic dystrophy type I. Genes 11:1109. https://doi.org/10.3390/genes11091109
Beqqali A, Bollen IAE, Rasmussen TB et al (2016) A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc Res 112:452–463. https://doi.org/10.1093/cvr/cvw192
Sedaghat-Hamedani F, Haas J, Zhu F et al (2017) Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur Heart J 38:3449–3460. https://doi.org/10.1093/eurheartj/ehx545
Khan MAF, Reckman YJ, Aufiero S et al (2016) RBM20 regulates circular RNA production from the titin gene. Circ Res 119:996–1003. https://doi.org/10.1161/CIRCRESAHA.116.309568
Methawasin M, Hutchinson KR, Lee E-J et al (2014) Experimentally increasing titin compliance in a novel mouse model attenuates the frank-starling mechanism but has a beneficial effect on diastole. Circulation 129:1924–1936. https://doi.org/10.1161/CIRCULATIONAHA.113.005610
Guo W, Zhu C, Yin Z et al (2018) Splicing factor RBM20 regulates transcriptional network of titin associated and calcium handling genes in the heart. Int J Biol Sci 14:369–380. https://doi.org/10.7150/ijbs.24117
van den Hoogenhof MMG, Beqqali A, Amin AS et al (2018) RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 138:1330–1342. https://doi.org/10.1161/CIRCULATIONAHA.117.031947
Schneider JW, Oommen S, Qureshi MY et al (2020) Dysregulated ribonucleoprotein granules promote cardiomyopathy in RBM20 gene-edited pigs. Nat Med 26:1788–1800. https://doi.org/10.1038/s41591-020-1087-x
Guo W, Zhu C, Yin Z et al (2021) The ryanodine receptor stabilizer S107 ameliorates contractility of adult Rbm20 knockout rat cardiomyocytes. Physiol Rep 9. https://doi.org/10.14814/phy2.15011
Wang C, Zhang Y, Methawasin M et al (2022) RBM20S639G mutation is a high genetic risk factor for premature death through RNA-protein condensates. J Mol Cell Cardiol 165:115–129. https://doi.org/10.1016/j.yjmcc.2022.01.004
Xu X, Yang D, Ding J-H et al (2005) ASF/SF2-regulated CaMKIIδ alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 120:59–72. https://doi.org/10.1016/j.cell.2004.11.036
Rossi D, Gamberucci A, Pierantozzi E et al (2021) Calsequestrin, a key protein in striated muscle health and disease. J Muscle Res Cell Motil 42:267–279. https://doi.org/10.1007/s10974-020-09583-6
Refaat MM, Lubitz SA, Makino S et al (2012) Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm 9:390–396. https://doi.org/10.1016/j.hrthm.2011.10.016
Wells QS, Becker JR, Su YR et al (2013) Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ Cardiovasc Genet 6:317–326. https://doi.org/10.1161/CIRCGENETICS.113.000011
Hey TM, Rasmussen TB, Madsen T et al (2019) Pathogenic RBM20 -variants are associated with a severe disease expression in male patients with dilated cardiomyopathy. Circ Heart Fail. https://doi.org/10.1161/CIRCHEARTFAILURE.118.005700
Parikh VN, Caleshu C, Reuter C et al (2019) Regional variation in RBM20 causes a highly penetrant arrhythmogenic cardiomyopathy. Circ Heart Fail. https://doi.org/10.1161/CIRCHEARTFAILURE.118.005371
Vakhrushev Y, Kozyreva A, Semenov A et al (2021) RBM20-associated ventricular arrhythmias in a patient with structurally normal heart. Genes 12:94. https://doi.org/10.3390/genes12010094
Yamashita Y, Matsuura T, Kurosaki T et al (2014) LDB3 splicing abnormalities are specific to skeletal muscles of patients with myotonic dystrophy type 1 and alter its PKC binding affinity. Neurobiol Dis 69:200–205. https://doi.org/10.1016/j.nbd.2014.05.026
Acknowledgements
The authors would like to thank the Joint-Use Research facilities and the Center for Comparative Medicine, Hyogo Medical University, for their technical assistance. The authors would also like to thank Dr. S. Nomura for his genetic advice.
Funding
This work was supported in part by grants from JSPS KAKENHI (JP20H03958 to T.Y., JP20K07659 and JP20K21385 to H.K.) and Takeda Science Foundation.
Author information
Authors and Affiliations
Contributions
T.Y., R.S., K.I., H.K., and H.N. conceived the study and experimental design. R.S. and Y.K. performed the genetic analysis and reporter assay experiment. M.I. and M.O. generated the knock-in mouse, and T.Y. and A.M. performed RNA analysis. T.Y., A.M., and Y.N. analyzed cardiac function. T.Y., M.N., and N.O. drafted the manuscript. All authors reviewed the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All research was performed in accordance with the guidelines for human genome analysis in Japan, and the study protocol was approved by the Ethics Committee of Hyogo Medical University (0358) and Gunma University Graduate School of Medicine (2017–015). All animal experiments were performed with the approval of the Hyogo Medical University Institutional Animal Care and Use Committee (18–078) and the ARRIVE guideline.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamamoto, T., Sano, R., Miura, A. et al. I536T variant of RBM20 affects splicing of cardiac structural proteins that are causative for developing dilated cardiomyopathy. J Mol Med 100, 1741–1754 (2022). https://doi.org/10.1007/s00109-022-02262-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-022-02262-8