
Abstract
Human sterile α motif and HD domain-containing protein 1 (SAMHD1), originally described as the major cellular deoxyribonucleoside triphosphate triphosphohydrolase (dNTPase) balancing the intracellular deoxynucleotide (dNTP) pool, has come recently into focus of cancer research. As outlined in this review, SAMHD1 has been reported to be mutated in a variety of cancer types and the expression of SAMHD1 is dysregulated in many cancers. Therefore, SAMHD1 is regarded as a tumor suppressor in certain tumors. Moreover, it has been proposed that SAMHD1 might fulfill the requirements of a driver gene in tumor development or might promote a so-called mutator phenotype. Besides its role as a dNTPase, several novel cellular functions of SAMHD1 have come to light only recently, including a role as negative regulator of innate immune responses and as facilitator of DNA end resection during DNA replication and repair. Therefore, SAMHD1 can be placed at the crossroads of various cellular processes. The present review summarizes the negative role of SAMHD1 in chemotherapy sensitivity, highlights reported SAMHD1 mutations found in various cancer types, and aims to discuss functional consequences as well as underlying mechanisms of SAMHD1 dysregulation potentially involved in cancer development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
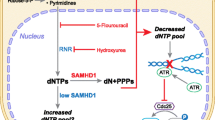
Human sterile α motif and HD domain-containing protein 1 (SAMHD1) was first described to be the major cellular deoxyribonucleoside triphosphate triphosphohydrolase (dNTPase) and to be crucial for controlling cellular deoxynucleotide (dNTP) levels [1, 2]. At present, the role of SAMHD1 in a variety of cancer types has been studied and will be highlighted in this review. Besides its role as a dNTPase, several novel functions have been attributed to SAMHD1. These include a direct role of SAMHD1 as a negative regulator of innate immunity [3], and a role in promoting the end resection process during DNA repair by recruitment of CtBP-interacting protein (CtIP) endonuclease to DNA damage sites [4] and during DNA replication by resolving stalled replication forks through recruitment of MRE11 Homolog, Double Strand Break Repair Nuclease (MRE11) and stimulating its exonuclease activity [5] (Fig. 1).

SAMHD1, its functions, and implications for AGS and cancer
Cellular functions of SAMHD1 and functional consequences for mutated/dysfunctional or downregulated SAMHD1 are depicted. Mutated SAMHD1 might lead to displacement of ssDNA into the cytoplasm, where it can be detected by intracellular DNA sensors like cGAS. cGAS then produces cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) to activate STING which in turn activates interferon regulatory factor 3 (IRF3) and the NF-κB pathways through the kinases TANK-binding kinase 1 (TBK1) and IκB kinase (IKK), thus inducing an IFN response. Consequences of dysfunctional SAMHD1 on AGS and cancer are displayed in the lower part of the figure. Unclear relations and consequences are indicated by question marks. Image created with Servier Medical Art (https://smart.servier.com/)
Furthermore, described mutations in SAMHD1 can cause the hereditary encephalopathy and interferonopathy Aicardi-Goutières syndrome (AGS) [6]. The exact mechanism of how mutated or inactive SAMHD1 triggers a type I interferon-mediated response is not yet clear. It is hypothesized to result from accumulation of self-derived nucleic acids, which trigger this response (Fig. 1) [7]. The source of the endogenous nucleic acids is as yet unclear; however, the various functions of SAMHD1, when inactive, could promote their accumulation and possibly lead to tumor-promoting inflammation (Fig. 1). It is known that unresolved DNA damage could lead to release of aberrant DNA into the cytosol, thus stimulating the cytosolic DNA sensor cyclic GMP-AMP Synthase (cGAS) and its adaptor Stimulator of Interferon Genes (STING) [8]. Therefore, mutations in SAMHD1 affecting DNA repair may lead to subsequent interferon (IFN) activation. Furthermore, mutations impairing end resection processes during DNA replication could lead to accumulation of aberrant DNA dislocated into the cytoplasm. Coquel et al. proposed that this aberrant DNA could activate innate immune signaling by cGAS/STING [5]. Also, mutations that impair the negative role of SAMHD1 within the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and Interferon regulatory factor 7 (IRF7) pathway might also come into play [3].
SAMHD1 is placed at the crossroads of various cellular processes, including cell cycle progression and proliferation. Whether SAMHD1 deficiency affects cell proliferation, however, is still under debate. In transformed cells, contrasting results were reported: SAMHD1 deficiency was shown to lead to reduced cell growth and altered replication dynamics [5, 9]. On the other hand, it was shown that SAMHD1 deficiency led to stimulation of cell proliferation and reduced spontaneous apoptosis induction [10]. Moreover, mutated or downregulated SAMHD1 could lead to improperly regulated nucleotide metabolism as well as malfunctioning DNA replication and repair processes which will potentially lead to genomic instability and accumulation of mutations (Fig. 1). Together with resistance to apoptosis [10], SAMHD1 is involved in several cellular processes which are important hallmarks of cancer when dysregulated, as defined and summarized by Hanahan and Weinberg (2011). SAMHD1 might influence a variety of hallmarks, potentially including tumor-promoting inflammation as an enabling characteristic in neoplastic disease [11]. Therefore its role in the development in different cancer types remains to be firmly investigated. Furthermore, it will be important to dissect the influence of the dNTPase function of SAMHD1 on the effects observed in cancer cells, or whether additional functions contribute to tumorigenesis. This will enable a better understanding of SAMHD1 as a target for cancer therapy.
SAMHD1 mutations reported in various cancer types
To investigate the mutation spectrum of SAMHD1 in cancer, we queried the International Cancer Genome Consortium (ICGC) database [12]. We found 1542 mutations of SAMHD1 affecting 957 donors across 65 cancer projects. Figure 2a illustrates the extent to which mutations occur in each cancer type. A high prevalence of mutations in Fig. 2a is due to intronic mutations and mutations within the 5′ and 3′ UTR. As we were interested in mutations likely to change the protein function, we calculated the percentage of donors affected per cancer type for only coding mutations (Fig. 2b). Missense mutations are more represented than all others among the coding mutations (Supplemental Table 1). The five most prevalent cancer types affected by coding mutations are endometrial, thyroid, skin, colon and liver cancer (Fig. 2b). Additionally, SAMHD1 was identified to be recurrently mutated in certain hematological malignancies (Fig. 2, “blood” cancer) and analyzed in detail as outlined in the next paragraph.

Donors affected by mutations in SAMHD1 per cancer type
The distribution of all (a) and only coding somatic mutations (b) across the 20 most prevalent ICGC cancer studies is represented. The ICGC data portal offers clinical and analyzed data representing 81 cancer type datasets available from the ICGC Data Coordination Center for Release 28 (human genome hg19/GRCh37), processed as of March 27, 2019. We used open-access simple somatic mutations (SSM) calls. These include single and multiple base substitutions, and small (≤ 200 bp) insertions and deletions that appear in the tumor tissue, but not in the normal control tissues. The figure legends in a and b depict all surveyed cancers that are included in the pie charts along with the calculated percentage (%) of donors affected by each cancer type
Chronic lymphocytic leukemia (CLL), the most frequent type of leukemia in adults, is characterized by heterogeneous and constantly changing cell populations, leading to complications like treatment relapse or resistance to chemotherapy [13]. Using whole-genome sequencing (WGS), Schuh et al. monitored shifts in tumor subclone populations in three patients over the course of CLL treatment. In one patient, a somatic mutation in SAMHD1 (c.1635 T > A; aa exchange: F545L) was identified in the founder subclone and present during all time points examined, indicating this mutation to be an early, potentially driving event [14]. Underscoring the role of SAMHD1 mutations in CLL, a patient carrying a homozygous germ-line mutation in SAMHD1 (c.1609-1G > C) was described in a subsequent study who was diagnosed with CLL at only 24 years of age — with no other acquired mutations or chromosomal lesions detectable known to be recurrently found in CLL [15]. Further analysis of clinical trial samples revealed that SAMHD1 mutations were present in 3% (pretreatment group) to 11% (relapsed/refractory group) of CLL patients [15]. Additionally, pre-existing subclones with mutations in SAMHD1 were enriched after therapy in another cohort of relapsed/refractory CLL (rCLL) patients, identifying genomic changes in SAMHD1 as possible drivers of relapse [16]. The authors even hypothesize that SAMHD1 mutations might contribute, to a certain extent, to CLL treatment resistance in vivo [16]. Furthermore, rCLL patients with > 1 gene mutation in nine recurrently affected genes (including SAMHD1 + either ATM/SF3B1/NOTCH1) showed significantly poorer outcome in terms of overall survival (OS) compared to patients with no or only one mutation [17]. Additionally, it is important to further understand mutational differences in CLL subtypes, which differ in the abundance of somatic hypermutations affecting the Ig variable heavy-chain locus (IgHVmut and IgHVunmut), since IgHVunmut patients display a more aggressive form of CLL with poorer OS compared to IgHVmut patients [18]. Burns et al. showed that mutations in the coding region and regulatory elements of SAMHD1, as well as in other known and potential CLL driver genes, were predominant in IgHVunmut CLL patients [19]. In future studies, it will be important to understand the effects of SAMHD1 mutations, especially in combination with other mutated genes, on the clinical outcome of different CLL patient groups in more detail.
As described, initial indications that SAMHD1 mutations might be involved in cancer development/progression were derived from the occurrence of early-onset CLL in an AGS patient [15]. By now, the case study of a patient initially diagnosed with SAMS (stroke, aneurysm, moyamoya, and stenosis) association, attributed to a homozygous mutation (c.1411-2A > G) affecting a splice acceptor site in SAMHD1 [20], was described who later developed a CD8+ epidermotropic cutaneous T-cell lymphoma (CTLC) at the age of 29 [21].
Apart from being recurrently mutated in rCLL, SAMHD1 mutations were also found in 18% of patients with T-cell prolymphocytic leukemia (T-PLL) [22]. In this study, SAMHD1 was identified as the second most frequently mutated gene, after ATM, and several mutations even presented as homozygous or hemizygous [22]. In T-PLL, some SAMHD1 mutations resulted in reduced mRNA expression; however, protein expression was reduced or even absent in all samples from T-PLL patients with SAMHD1 mutations [22]. Only recently, mutations in SAMHD1 were also detected in 7.1% (13/182) of mantle cell lymphoma (MCL) patients selected from the MCL Younger and Elderly trials; of note, both cohorts only included previously untreated patients [23]. MCL is an rare subtype of B-cell non-Hodgkin lymphoma, which shows an aggressive course of disease and is still considered incurable [24]. In mutated MCL cases, SAMHD1 protein expression decreased compared to SAMHD1-unmutated patients, although the difference did not reach significance [23]. However, the mutation status of SAMHD1 had no significant influence on failure-free survival (FFS) of MCL patients [23].
By now, a single case of an extremely rare low-grade B-cell lymphoma with IRF4 rearrangement with concomitant mutation in SAMHD1 (c.G692A; leading to premature a stop codon, W231X) was described [25]. Probably, this early stop-gain mutation in SAMHD1 will lead to reduced protein expression, which was not assessed in the report by Zhou et al. In this patient, further missense and frameshift mutations were detected in the genes KMT2D, BTG1, PTEN, and BAX [25].
As SAMHD1 mutations were identified in several hematological malignancies, future efforts will be important to investigate whether SAMHD1 mutations can be found in other tumors of hematopoietic and lymphoid origin and to further pinpoint which exact amino acid residues in SAMHD1 are affected, in order to investigate their roles in SAMHD1’s diverse cellular functions. Importantly, changes in SAMHD1 protein functions and/or expression levels due to deleterious mutations can have significant influence on the therapeutic outcome of specific cancer treatments (see last section of this review).
Apart from hematological malignancies, first studies on the role of SAMHD1 mutations in the development and progression of solid tumors were conducted only in recent years. Using the colorectal cancer (CRC) data set deposited in The Cancer Genome Atlas (TCGA), Rentoft et al. identified eight different, nonsynonymous mutations in the coding region of SAMHD1; here, the amount of mutations in SAMHD1 was higher than expected by chance [26]. Interestingly, all eight mutations were found in hypermutated colon cancers (> 12 mutations per 106 bases) — with six of these tumors carrying additional mutations in genes important for mismatch repair (MMR). In S. cerevisiae, even a minor elevation in dNTP concentrations, in combination with mutated MMR genes, can lead to reduced DNA replication fidelity and, as a consequence, increased mutation rates [26]. It will be of great interest to study a possible interplay of SAMDH1 and MMR defects in mammalian cells, in general, and specifically in different tumor types. In vitro characterization of selected CRC-associated SAMHD1 mutants (V133I, A338T, R366H, D497Y) (Table 1) revealed that, indeed, all of them showed reduced or even completely abolished dNTPase activity compared to wild-type (wt) SAMHD1 [26]. In addition, some mutations (like R366H) did not influence hydrolysis of individual dNTPs to the same extent (2.5- to 11-fold for deoxyadenosine triphosphate/deoxycytidine triphosphate/deoxythymidine triphosphate (dATP/dCTP/dTTP), almost no effect on deoxyguanosine triphosphate (dGTP)), indicating that not only absolute, but also relative dNTP levels could be influenced by SAMHD1 mutations [26]. Using hemizygous SAMHD1+/− mouse embryos, Rentoft et al. could show that inactivation of only one SAMHD1 allele leads to elevation of cellular dNTPs. Consequently, the authors speculated that heterozygous, inactivating SAMHD1 mutations would also disturb dNTP pools in vivo. However, in future studies, the exact impact of (heterozygous) SAMHD1 mutations on dNTP levels/balance and/or mutation rates needs to be addressed using primary CRC patient samples.
In summary, it will be important to address how acquired SAMHD1 mutations provide an advantage for cancerous cells and whether differences between tumor types are observable (for instance, hematological malignancies vs solid tumors). In general, it will be of great interest to understand how the different cellular roles of SAMHD1 (like dNTP homeostasis or involvement in DNA replication/DNA damage response (DDR)) are potentially disturbed through mutations, thereby likely driving oncogenesis.
SAMHD1 mutations and their functional significance
SAMHD1 is involved in controlling absolute and relative cellular dNTP levels [2] and capable of degrading all four dNTPs [1, 27]. Therefore, functional mutations in SAMHD1 could lead to dNTP imbalances. Consequently, disturbed DNA replication fidelity along with spontaneous mutations could result in genomic instability, potentially promoting cancer development [28].
The regulation of dNTP pools is important for cell cycle progression, as cycling cells need to carefully balance dNTP levels to ensure proper S phase completion and transition to mitosis [29]. During G1 phase, SAMHD1 maintains low dNTP levels. Only upon entering into S phase, the dNTPase activity of SAMHD1 is potentially downregulated through phosphorylation at residue T592 [30,31,32] and/or reduction of its protein level [2]. Cancer cells need to sustain chronic proliferation; therefore, they need high dNTP levels at all times [33]. This could be achieved by downregulating SAMHD1 expression (see Fig. 3) or through the acquisition of mutations in SAMHD1 which abolish its dNTPase activity.

Expression of SAMHD1 in different cancer types
Each point represents paired tumor/healthy samples and the relative difference of SAMHD1 expression between the two. The difference is represented as a z-score, which shows the number of standard deviations between the expression of SAMHD1 in the respective tumor sample and the mean expression of SAMHD1 in the healthy samples
We visualized the expression of genes originating from the resource PanCancer Atlas [34] by using cbioportal [35, 36]. Interestingly, SAMHD1 displays a general downregulation in most cancer types, implying a correlation between cancer and SAMHD1 repression — with the strongest downregulation observed in lung cancers (Fig. 3). Next, we were interested how coding mutations are distributed throughout the protein sequence of SAMHD1. The lollipop diagram in Fig. 4 illustrates a graphical representation of the somatic mutation spectrum of SAMHD1. All 230 coding mutations seem to distribute relatively evenly throughout the whole protein sequence of SAMHD1 (Fig. 4, Supplemental Table 1), which, at first glance, does not allow us to draw any conclusions on certain protein domains that might be important for cancer development. This also suggests that not only the enzymatic activity of SAMHD1 might be responsible for tumor development.

Graphical representation of the somatic mutation spectrum throughout the protein sequence of SAMHD1
In total, 177 coding mutations from ICGC cancer studies and other 53 mutations surveyed from the literature were visualized. The scale bar represents the length (amino acids) of the protein sequence. Each lollipop represents a somatic coding mutation. Lollipops are colored according to the consequence type: missense (red), frameshift (blue), stop-gain (purple), stop-lost (olive), deletion (yellow). The size of the lollipops represents the number of reported patients with the mutation. The lollipop diagram was created by using [106]. The domain structure is based on [100]. Supplemental Table 1 lists the 230 coding mutations inclusive cancer type and references
In the next chapter, we will discuss the impact of cancer-associated mutations on specific functions of SAMHD1. As SAMHD1 is expressed to different levels in different tissues and cancers, the impact of SAMHD1 might vary depending on the tissue in question. Moreover, the diverse functions of SAMHD1 make it difficult to pinpoint the exact mechanisms how SAMHD1 contributes to tumor development. Furthermore, partial or complete loss of SAMHD1 expression could be caused by specific mutations. This can be observed in CLL where many patients show reduced or abolished SAMHD1 expression due to somatic mutations [15]. Therefore, in this review, our aim is to provide a detailed overview of the cancers that are affected by SAMHD1 and to summarize the reported mutations and corresponding functional consequences. The most interesting mutations that either have reported known functional consequences or a known involvement of the respective amino acid in structural integrity/cellular functions are listed in Table 1.
Known and potential impact of cancer-associated mutations on SAMHD1 function
Changes in structure/catalytic function
The functionality of SAMHD1’s dNTPase activity is dependent on the catalytic and the allosteric sites in the HD domain of SAMHD1 [1]. Therefore, mutations in this region of the protein can reduce or completely abrogate the dNTPase function. Goldstone et al. created a panel of catalytic and allosteric site mutants that show such effects. All reported positions (H206A/D207A, D311A, H233A, R164A, D137A, Q142A, R145Q) can be found mutated in cancer patients (see Table 1; Fig. 4, Suppl. Table 1). This indicates that mutations that interfere with dNTPase function might lead to dNTP pool imbalances in these patients that could cause genomic instability or a mutator phenotype. Additionally, mutations associated with colon adenocarcinoma (V133I, A338T, R266H, and D497Y) (Table 1) were shown to reduce the dNTPase activity of SAMHD1 (see previous section). The resulting dNTP pool imbalances caused an increase of mutation frequency, when combined with MMR deficiency [26]. Furthermore, some catalytic or allosteric site mutants are associated with AGS (residues H123; R143, R145, R164, H167, R333, M385, and Q548) (Table 1) and might therefore lead to the induction of IFN [6, 37]. These residues were also reported in cancer patients. Other catalytic and allosteric site mutations which can be found in cancer involve residues D137, Q142, H206, D207, H233, D311, R366, and R451 (Table 1).
The relationship of dNTP pool balance and genomic instability was primarily shown for the ribonucleotide reductase, the rate-limiting enzyme of the de novo dNTP pathway [33]. Consequently, also upregulation or downregulation of SAMHD1 expression or SAMHD1 mutations, which might alter its dNTPase activity, can contribute in a comparable manner [29, 38]. Dysregulated dNTP levels can be responsible for DNA replication stress and can affect DNA repair mechanisms [29]. Additionally, increased frequencies of DNA damage by dNTP pool dysregulation can induce IFN-stimulated genes, leading to chronic inflammation, a phenotype commonly observed in AGS patients [6, 38]. Another consequence of dNTP pool imbalances can be the increase of random genome-wide mutations, thus creating a mutator-phenotype, driving oncogenic transformation of pre-cancerous cells [39].
SAMHD1 has also been proposed to play a role during antibody class switch recombination [40]. During this process, non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ) are active and reported to be sensitive to dNTP imbalances caused by dNTPase-impaired SAMHD1. This could lead to nucleotide insertions at the recombination sites leading to genomic instability in B cells [41].
Mislocalization
SAMHD1 is primarily localized in the nucleus due to the nuclear localization sequence 11KRPR14 [42]. Mutations that lead to changes in SAMHD1 localization have been identified in AGS patients and might therefore contribute to pathogenicity [43]. These mutations can also be found in cancer, especially CLL [15]. Mutations that are common in AGS and cancer are H123Y, R143H/C, R145X/Q, R164Q, G209C, I201N, R226H, M254I, R290C, D311E, R442X, and I448T (Table 1, also includes other amino acid changes at the same amino acid residue) [37, 43]. Interestingly, many of these AGS mutations were reported for CLL: R145X/Q, I201N, R290C, and M254I [15, 44]. Except for the D311 mutation, all of these mutants show mislocalization to the cytoplasm to different degrees [37]. This indicates that mislocalization of SAMHD1 might not only contribute to pathogenicity in AGS but also in certain cancer types, namely CLL. This phenomenon speaks for a role of SAMHD1 in replication and/or DNA damage response as nuclear localization might not be essential for dNTPase activity. However, mutations at R143, I201, R226, M254, D311, R442, and I448 were shown to have lost dNTPase activity [37, 45]. Therefore, future studies are needed to determine to which extent SAMHD1 mislocalization might influence or even obstruct its cellular activities. As these mutations are all reported AGS mutations, they might all lead to an upregulation of interferons as the typical AGS phenotype described by Rice et al. [46] although this remains to be conclusively validated.
Dimer and tetramer formation
dNTPase-active SAMHD1 forms tetramers [47,48,49]. Phosphorylation at T592 downregulates the dNTPase function of SAMHD1 by interfering with protein tetramer stability [31, 32, 50]; however, conflicting results have been reported, de-coupling phosphorylation and dNTPase activity [51]. Phosphorylation of SAMHD1 is regulated during cell cycle progression [30, 31] and occurs in cycling cells during cell cycle phases that require high dNTP concentrations, i.e., during DNA replication in S phase. In terminally differentiated cells, SAMHD1 is usually not phosphorylated at residue T592 to maintain low dNTP pools [30]. It could be plausible that in certain cancers, SAMHD1 remains phosphorylated, as cancer cells are metabolically highly active with reduced or lost control mechanisms for cell growth. This can be observed in cultured cancer cell lines, e.g., cycling THP-1 cells [51]. Interestingly, so far, T592 mutations have not been reported to occur in cancers (see Table 1; Fig. 4, Suppl. Table 1). This could indicate that it is beneficial for cancers to maintain SAMHD1 phosphorylation. However, as it yet remains to be clarified how T592 phosphorylation is connected to tetramerisation and dNTPase function [52], one can only speculate on the influence of SAMHD1 T592 phosphorylation on cancer cells. In this context, SAMHD1 mutations that affect dimerization or tetramerization ability might play a role as well. Intriguingly, we could find two reported mutations (H364Q and H364K) deposited to ICGC which might affect SAMHD1 tetramerization [53].
DNA replication and DNA end resection
Mutated SAMHD1 could not only contribute to genomic instability through an impaired dNTPase activity but also due to its role in end resection during DNA replication or DNA damage repair [4, 5]. In the absence of SAMHD1, end resection, a process necessary to resolve stalled replication forks and enable the repair of DNA double strand breaks, is not functioning properly [5]. Coquel et al. could show an interaction of SAMHD1 and MRE11 nuclease, stimulating the exonuclease function of this enzyme. This activity initiates DNA end resection and consequently downstream processes to activate DNA damage repair and replication fork restart [7]. Daddacha et al. made similar observations for CtIP, another enzyme involved in initiating the DNA end resection process, which also interacts with MRE11, linking SAMHD1 to the initiating events of homologous recombination (HR) [4]. In both reports, SAMHD1 facilitates recruitment of the factors to enable end resection.
Without end resection, cells are not able to replicate correctly, which could lead to accumulation of genomic mutations, thus contributing to genomic instability and possibly to mutation of proto-onco genes.
Involvement of SAMHD1 in end resection could provide a link to the reported ability of SAMHD1 to bind nucleic acids, like single-stranded DNA (ssDNA). This has been implicated in several publications [45, 54, 55], and although it is not clear whether SAMHD1 itself can act as a nuclease, it seems plausible that it can bind ssDNA and recruit nuclear endonucleases or exonucleases which can then degrade nucleic acids [55]. Interestingly, amino acid residues that were characterized by Seamon et al. to be involved in ssDNA binding overlap with cancer-associated mutations in the region between residue 360 and residue 545: Y360, H364, Y521, F545 (Fig. 4, Suppl. Table 1). These residues are not part of the active or allosteric site of SAMHD1. This might indicate that mutations in this region specifically alter ssDNA binding ability of SAMHD1, while other functions are not affected. It would be interesting to study these mutants with regard to their ability to promote DNA end resection by recruiting MRE11 exonuclease [5]. Another mutation that can be found in this protein region is the K484T mutant described by Daddacha et al. who have shown that this residue is important for the recruitment of CtIP to DNA damage sites to enable DNA end resection in HR [4]. Both these studies provide mechanistic insight how SAMHD1 plays a role in replication and DNA damage and how its ssDNA binding ability might be connected to this function. Interestingly it was also shown in both publications that the involvement of SAMHD1 in DNA end resection is dNTPase-independent. dNTPase-defective H206A/D207A [4] and K312A [5] were still able to induce the SAMHD1 wt phenotype in rescue experiments, while the partially dNTPase-active Y315A [45] mutant could not rescue the wt replication phenotype [5]. In contrast, the T592 phosphorylation seems to play a role in this regard, as the phosphoablative mutant T592A can no longer rescue the WT phenotype, while the phosphomimetic T592E mutant can [5]. This highlights that the T592 phosphosite might serve as a switch of function between controlling viral restriction and end resection, regulated during the cell cycle. During S-phase, phosphorylated SAMHD1 (at T592) supports the replication process, while it is rapidly dephosphorylated at T592 during mitotic exit and restrictive towards HIV-1. Mitotic exit might also mark the transition to its function as a dNTPase controlling the cellular dNTP pool [5, 30, 50].
Recently, another aspect emerged. At DNA replication-transcription conflict regions, R-loops (DNA:RNA hybrids) are formed and are associated with cancer development [56]. These are enriched in AGS patients with SAMHD1 deficiency and can cause replication stress and genome instability [57]. SAMHD1 was shown to be involved in resolving R-loops and it was suggested that this might also be connected to its ability to recruit MRE11 [57]. In this study, colorectal cancer-associated mutations (F59C, D207Y, R226H, T232M, K288T, and S247Y [58] (Fig. 4, Suppl. Table 1)) were investigated on their ability of regulating R-loops. Two of these mutations (F59C and T232M) showed increased transcription-replication conflicts when compared to SAMHD1 wt [57]. This provides another likely dNTPase-independent mechanism of how SAMHD1 could be involved in cancer.
End joining
Recently, SAMHD1 was shown to be also involved in DNA end joining in NHEJ, a DNA repair pathway active throughout all cell cycle phases [59]. It was described that the dNTPase function of SAMHD1 is important for this repair pathway as a balanced dNTP pool is necessary to avoid nucleotide insertions at repair junctions, potentially linking SAMHD1 dysfunction to genomic instability and thereby promoting tumor development [60]. This was also observed during antibody class switching [40, 41]. This is especially intriguing, as SAMHD1 is frequently mutated in CLL, a form of B lymphocyte leukemia. This model provides an explanation for one specific subset of cells (B cells) and is of course not applicable for the majority of other tumors. In this context, the dNTPase defective K312A mutation leads to longer repair junctions harboring DNA insertions. In contrast, mutations K484T and K11A with intact dNTPase activity did not lead to longer DNA insertions [60]. A mutation at K484 is also found in the cancer data (Table 1; Fig. 4, Suppl. Table 1). This suggests that SAMHD1 might play different roles in different tumors, depending on tissue-specific factors, like expression, cell division rates, and specialization of a cell type.
Innate signaling
Mutated SAMHD1 is strongly connected to inflammation. In the absence of functional SAMHD1, cells are not able to control upregulation of inflammatory signals. This can be observed in the hereditary autoimmune disease AGS, in which patients display chronically elevated IFN levels [6]. In this context, IFN stimulatory self-DNA has been implicated as a possible cause for the disease phenotype [61]. However, the exact reasons are not yet fully understood. On the one hand, SAMHD1 itself can act as a negative regulator of innate immunity [3]. On the other hand, SAMHD1 prevents aberrant DNA being dislocated into the cytoplasm by helping to resolve stalled replication forks [5]. As displayed in Fig. 1, SAMHD1 role in DNA repair might play an additional role in upregulation of innate immunity [8]. A similar concept might apply for cancer development as well. One could hypothesize that SAMHD1, if mutated or downregulated in cancer cells, would lead to accumulating self-DNA that might be sensed through the cGAS-STING pathway thus inducing a tumor-associated chronic inflammatory response [7, 62]. Therefore, SAMHD1 might be placed into the group of caretaker genes that protect cells from genomic instability [63] by reducing DNA damage and thereby potentially avoiding the induction of a strong immune response by self-DNA [38].
Tumors are often infiltrated by immune cells, both of the innate and adaptive arms, resembling inflammatory conditions [11]. Of course, a strong immune reaction is crucial to destroy tumors. Paradoxically, immune cells, particularly innate immune cells, can also contribute to neoplastic progression by providing bioactive molecules to the tumors which can facilitate tumor growth [11]. One inflammatory cytokine which has been linked to pro-tumor effects is the tissue necrosis factor α (TNF-α) when produced in the tumor microenvironment. TNF-α is produced by various cancers in small quantities and can promote cancer progression in various ways which are still under investigation [64, 65]. SAMHD1 was shown to be a negative regulator of innate immunity. Specifically, it has been linked to the Nf-κB pathway. SAMHD1 interacts with the Nf-κB inhibitor IκBα by blocking the phosphorylation and subsequent degradation of this inhibitor. Additionally, SAMHD1 inhibits IκB kinase ε (IKKε)-mediated IRF7 phosphorylation and by this reduces IFN-1 induction [3]. Therefore, missing or mutated SAMHD1 might contribute to a pro-tumor microenvironment, as the cells are not able to correctly downregulate inflammatory signals like TNF-α in the absence of SAMHD1 [3].
Also the cGAS-STING pathway, which is known to have important implications in anti-tumor immunity [62], can in some cancers promote inflammation-driven carcinogenesis, for example in brain metastasis [66] or skin cancer [67]. It is thought that DNA leakage into the cytoplasm triggers cGAS-STING-dependent production of pro-inflammatory cytokines like TNF-α [67].
There is little known about SAMHD1 mutations that influence its ability to downregulate the NF-κB pathway. It was shown that dNTPase activity is important for this function, as the dNTPase-defective SAMHD1 mutant H206R/D207N loses the ability to downregulate innate signaling in nondividing monocytic cells [68]. Therefore, it is possible that other SAMHD1 mutants with reduced dNTPase activity, e.g., R305A, D311A, K312A, and R143H (Table 1, Fig. 4, Suppl. Table 1) [45], also lose this function and contribute to chronic inflammation associated with the tumor microenvironment. Interestingly, for cycling cells, the dNTPase activity is not involved in downregulating innate immunity, highlighting that SAMHD1 functions seem to be highly dependent on cell cycle status and cell proliferation activity [3]. Also, the phosphorylation status at T592 does not seem to contribute to SAMHD1’s role in regulating the innate immune response [3].
Known AGS mutations can be found in various cancer types: R145X, R143C, R442X, R145Q, R611Q, R348C, D585N, P485S, A181T, R194X, R339C, R333H (overlap of ICGC cancer mutations and AGS mutations). Many of these mutations also have functional significance (see above and Table 1). As depicted in Fig. 1, SAMHD1 could be involved in different cellular processes, which lead to the upregulation of the IFN signaling, also independent of its dNTPase activity. It was shown that AGS patient-derived fibroblasts with either R290H, Q548X, or H167Y mutations display an altered dNTP pool leading to genomic instability and upregulation of IFN [38]. The R290 residue is also represented in the cancer data survey (Table 1, Fig. 4, Suppl. Table 1). In conclusion, the impact of SAMHD1 on innate signaling and perhaps on additional functions might contribute to inflammation-driven carcinogenesis.
SAMHD1 as a potential tumor suppressor
Tumor suppressor genes are vital to regulate normal cell growth and proliferation. Therefore, their expression is repressed on the transcriptional level in various malignancies, for instance, through promoter methylation and/or histone modifications [69].
Initially, it was reported that SAMHD1 mRNA and protein expression were reduced in peripheral blood mononuclear cells (PBMCs) obtained from patients with Sézary syndrome (SS), an aggressive subtype of cutaneous T-cell lymphoma (CTLC), compared to healthy donors [70]. In eight out of nine patient PBMCs examined, the SAMHD1 promoter was highly methylated (up to 51-fold higher on average), whereas no promoter methylation could be observed in PBMCs from healthy donors [70]. Subsequent studies aiming to identify recurrently mutated/altered genes in SS patients found deletions or mutations in SAMHD1 [71], potentially leading to altered SAMHD1 expression, in > 10% of patients.
SAMHD1 downregulation on the mRNA and protein level, compared to CD4+ T-cells from healthy donors or monocytic THP-1 cells, was also observed in various CD4+ T-cell lines derived from leukemia and CTCL patients [72, 73]. Reduced SAMHD1 expression was achieved through transcriptional repression by promoter methylation [72], potentially in combination with microRNA-181 upregulation [73]. Specifically, an inverse correlation between miRNA-181b levels and SAMHD1 protein expression could be established [73]. Additionally, increased expression of all microRNA-181 family members (a-d) was detected in primary CD4+ T-cells from Sézary syndrome patients compared to healthy control cells, which was again associated with reduced SAMHD1 protein expression [73]. However, the exact contribution of both mechanisms to SAMHD1 downregulation, especially in CTCL patients (as, for instance, mRNA expression levels were in some patients reduced [70], while not in others [73]), would be interesting to explore in future studies. Nevertheless, to understand the impact of SAMHD1 downregulation in this cancer type, the CTCL-derived cell line HuT 78 (normally expressing low SAMHD1 levels) was stably transduced with full-length SAMHD1. As a result, reduced cell proliferation and colony formation, but higher levels of spontaneous and Fas ligand (FasL)-induced apoptosis were observed [74]. Therefore, it was proposed that SAMHD1 might act as a tumor suppressor in neoplastic T-cells partly by apoptosis induction [74].
As described, mutations in SAMHD1 were recurrently found in CLL patients leading to reduced mRNA expression and in most cases, but not all, almost complete loss of SAMHD1 protein expression compared to B-cells (mRNA) or PBMCs (protein) from healthy donors [15]. However, the exact mechanisms of mRNA/protein downregulation still need to be assessed in more detail. For instance, mutations could either interfere with proper transcription, induce non-sense mediated mRNA decay, or, in the end, could destabilize SAMHD1 (mutant) protein. Additionally, 12 out of 18 SAMHD1-mutated CLL patients showed abnormalities involving the SAMHD1 locus, located on chromosome 20, including copy-neutral loss of heterozygosity (cnLOH), mosaic cnLOH, or even complete loss of the second allele [15].
In T-PLL, SAMHD1 is not only recurrently mutated but deletions in the SAMHD1 locus were additionally observed in two patients (2/14 patients, 14%), resulting in completely abolished SAMHD1 protein expression [22]. As already noted in some CLL patients, a strict correlation between lower SAMHD1 mRNA levels and protein expression could also not be established for all T-PLL samples, again hinting at additional regulatory mechanisms. Additionally, the authors state that they could not detect hypermethylation of the SAMHD1 promoter in over 50 different T-PLL samples compared to healthy T-cells [22].
Acute lymphoblastic leukemia (ALL) is the most common cancer observed in children; due to improved treatment, survival rates have now increased from 10% in the 1960s to about 90% today [75]. Different subtypes of ALL might arise after malignant transformation of precursor cells from the B- or T-lymphoid lineage (B-ALL and T-ALL), characterized by specific alterations and gene expression patterns [75]. Rothenburger et al. were able to show specific differences in SAMHD1 mRNA expression between T- and B-ALL: In cell lines derived from T-ALL, SAMHD1 mRNA levels were significantly reduced compared to cell lines with B-ALL origin; the same mRNA expression pattern could also be detected in T- versus B-ALL blasts from 306 ALL patients [76]. Interestingly, a closer examination of SAMHD1 mRNA levels in T- and B-ALL subgroups revealed further pronounced differences, which could be important for therapeutic outcome; for instance, SAMHD1 mRNA levels were equally low in Philadelphia (Ph)-like B-ALL and T-ALL patient samples [76]. In T-ALL-derived cell lines, low SAMHD1 mRNA levels also correlated with low protein expression [76]. In order to understand the marked differences in SAMHD1 expression among T- and B-ALL-derived cell lines, investigation of SAMHD1 promoter methylation revealed that it was methylated in almost all T-ALL cell lines (10/11) tested, while being unmethylated in most B-ALL cell lines (13/15) [76]. Effectively, this observation suggests a lineage-specific regulation of SAMHD1 promoter methylation.
Acute myeloid leukemia (AML) is a hematological cancer that is characterized by uncontrolled proliferation of myeloid precursor cells in the bone marrow and blood, ultimately interfering with normal production of blood cells [77]. SAMHD1 mRNA expression levels differed widely in adult and pediatric AML patients [78]. As already observed in CTCL patients [70, 73], SAMHD1 mRNA expression negatively correlated with promoter methylation and levels of miRNA-181a [78]. With this, Herold et al. could define two distinct groups of AML patients (SAMHD1-low and -high expression) that respond differently to treatment with high-dose cytarabine (cytosine arabinoside, ara-C) consolidation therapy [78] (see last section in this review). Additionally, it was reported that SAMHD1 mRNA expression was downregulated in bone marrow samples of AML patients compared to a non-AML patient group [79]. SAMHD1 mRNA expression was not correlated with other downregulated apoptotic genes (BAD, BAX, BAK1, XIAP, and BIRC2) known to be relevant in AML pathogenesis. In this (small) AML cohort, however, low SAMHD1 mRNA expression was not associated with worse prognostic outcome (e.g., represented by white blood cell count or blast percentage) or reduced OS [79]. Nevertheless, a potential role of SAMHD1 as a tumor suppressor in AML still needs further investigation, as low SAMHD1 levels in AML bone marrow samples might indicate its role in leukemia induction. Investigating the potential involvement of different SAMHD1 functions, e.g., dNTPase, regulation of DNA replication/DDR, or inflammatory signaling in AML development and progression would be of great interest.
Only recently, SAMHD1 protein expression was assessed by immunohistochemistry in Hodgkin lymphoma (HL): Staining for SAMHD1 was mainly observed in the nucleus of Hodgkin and Reed-Sternberg (HRS) cells, which are the distinct neoplastic cells derived from mature B-cells found in HL. In total, only 31% (48/154) of HL samples evaluated were categorized as positive for SAMHD1 protein expression [80], implicating a potential downregulation of SAMHD1 in HL. The authors could also correlate SAMHD1 expression with clinical outcome in 125 HL patients: Here, positive SAMHD1 expression in HRS cells was linked to inferior freedom from progression (FFP; 51% vs 70%), disease-specific survival (DSS; 72% vs 92%), and 10-year OS (OS; 69% vs 86%) compared to HL patients with SAMHD1-low/negative HRS cells [80]. Therefore, SAMHD1 was suggested to be used as an independent marker for HL prognosis. It will be of great interest in future studies to determine how higher SAMHD1 expression can lead to poorer clinical outcome in HL.
Additionally, SAMHD1 expression seems to be downregulated, both on the mRNA and protein level, in malignant tissue from five lung adenocarcinoma (LAC) patients compared to the surrounding unaffected lung tissue [81] (see also Fig. 3). Again, the authors could correlate high SAMHD1 promoter methylation with lower SAMHD1 levels in lung adenocarcinoma [81]. To support the observations made in LAC patients, treatment of lung carcinoma-derived cell lines (A549, H1299) with the methyltransferase inhibitor 5-Aza-dC (decitabine) led to increased SAMHD1 mRNA and protein levels [81]. Combination of 5-Aza-dC with the histone deacetylase inhibitor trichostatin A (TSA) further increased SAMHD1 mRNA induction, indicating that several epigenetic mechanisms control SAMHD1 expression [81]. Mechanistically, overexpression of exogenous SAMDH1 in A549 cells led to reduced dNTP levels and cell proliferation compared to control cells [81]. Therefore, high SAMHD1 expression might confer a growth disadvantage for LAC cells.
In a subsequent study, SAMHD1 mRNA expression in tumor and adjacent healthy tissue was assessed in a larger cohort of 238 non-small lung cancer (NSLC) patients: Again, expression of SAMHD1 mRNA was significantly reduced in tumor compared to healthy specimens [82]. Interestingly, lower SAMHD1 mRNA expression correlated with a more aggressive, metastatic course of disease in patients [82]. To further understand SAMHD1’s involvement in lung cancer development and progression, A549 cells were stably transduced with SAMHD1 and reduced cell proliferation, colony formation, and apoptosis induction could be observed [82]. In line with the clinical data, SAMHD1 overexpression reduced A549 cell migration and invasion [82]. However, a potential negative regulation of STING by SAMHD1 and a resulting suppression of LAC progression, suggested by Wu et al., urgently need further experimental clarification [82].
According to our analysis, SAMHD1 expression can differ considerably between patients in lung adenocarcinoma (Fig. 3); therefore, it will be important to determine the exact impact of SAMHD1 downregulation in lung adenocarcinoma development and/or progression in future studies, especially the underlying molecular mechanisms.
The incidence of cutaneous melanoma (or skin cutaneous melanoma, SKCM), one of the most aggressive forms of skin cancer due to its potential to metastasize, is increasing annually and displaying high levels of somatic genetic alterations [83]. Using changes in DNA methylation profiles and copy number variations (CNVs) observed in SKCM, Chen et al. were able to define four distinct SKCM subtypes (iC1–iC4) that differed in their prognostic outcome — with the iC3 subtype showing the poorest OS [84]. Having a closer look at genes with distinct differences between subtypes (in terms of methylation status, CNV, and mRNA expression) revealed that 146 genes were actually correlated with prognosis [84]. Indeed, decreased mRNA expression and hypermethylation of SAMHD1 (along with GBP5, CD8A, and KIAA0040) were associated with reduced survival rate of patients clustered in the iC3 compared to the iC1 subtype [84].
A first analysis of primary breast cancer samples indicated that SAMHD1 protein expression might be reduced or even absent in approximately 50% of cases [15]. However, SAMHD1 protein expression was only compared to THP-1 and SupT1 cells, not to matched healthy breast tissue, and the reason for lower SAMHD1 expression (for instance, promoter methylation and/or detrimental mutations) will need further clarification. Additionally, it would be of great interest in further studies of breast cancer samples to discriminate between different subtypes and correlate whether differing SAMHD1 expression has an impact on disease prognosis.
Cancer-derived cell lines can be a viable tool to understand the molecular mechanisms leading to uncontrolled cell growth observed in malignant diseases. Specifically, it will be important to investigate the consequences of SAMHD1 deregulation (whether it is due to mutations or mRNA/protein downregulation) for cellular dNTP metabolism, DNA replication, and/or DDR which is necessary to evaluate SAMHD1’s role in cancer development and progression. The effect of SAMHD1 protein reduction or absence on cell proliferation, however, seems to differ depending on the cell type or cellular context. In a first study, reduction of SAMHD1 through RNA interference (RNAi) in immortalized cycling lung fibroblasts resulted in reduced cell growth, as G1/S transition during cell cycle-progression seemed to be disturbed. Consequently, cells accumulated in G1 phase, while the amount of S phase cells was reduced; however, no concomitant decline in dNTP concentrations could be observed [2]. Using SAMHD1-deficient primary fibroblasts from two different AGS patients, Kretschmer et al. could measure significantly upregulated dNTP levels, while the AGS fibroblasts also proliferated slower compared to healthy control cells [38]. In addition, AGS fibroblasts showed an overall decrease in genomic integrity and upregulation of several DDR genes [38].
CRISPR/Cas9-mediated knock-out (KO) of SAMHD1 in monocytic, AML-derived THP-1 cells resulted in increased cellular dNTP levels and an enrichment in G1/G0 phase-cells, while a reduction only in the G2/M population occurred [10]. In contrast to human fibroblasts [2, 38], these changes led to increased proliferation of THP-1_KO SAMHD1 compared to control cells [10].
By now, several studies also investigated the influence of SAMHD1 overexpression on proliferation in cancer-derived cell lines: In the cervical carcinoma-derived HeLa cells [15] and the lung cancer-derived cell line A549 [81], SAMHD1 overexpression led to reduced cell proliferation [15, 81] and a decrease in cellular dNTP levels [81]. Similar results were obtained using the CTCL-derived cell line HuT 78: SAMHD1 overexpression reduced dNTP concentration and cell proliferation, while apoptosis rates in these cells were elevated [74]. However, Herold et al. could not confirm the impact of SAMHD1 absence (in THP-1 cells) or overexpression (in HuT 78 cells) on cell proliferation [85]. Nevertheless, these conflicting results again highlight the importance of understanding the interplay of deregulated dNTP pools, DDR induction, and, ultimately, control of cellular proliferation due to changes in SAMHD1 activity. The contribution of SAMHD1’s different cellular functions will be important to elucidate — specifically, in various physiological and malignant cellular environments and particularly in primary cells.
In summary, SAMHD1’s potential role as a tumor suppressor is underlined by various studies and our own analyses (Fig. 3) that show its downregulation or deregulation through mutations in malignant diseases. Additionally, in many cancers, high expression levels of SAMHD1 are associated with a more favorable outcome (Table 2). Ultimately, SAMHD1 is required to balance cellular dNTP concentrations and/or regulate DNA repair/replication and, therefore, avoid mutagenic conditions favorable for cancer development and progression.
SAMHD1 as a potential driver gene
During the last 15 years, the genomic/mutational landscape of different human malignancies was extensively explored using large-scale sequencing efforts. Various genetic aberrations (from somatic mutations, to chromosomal as well as epigenetic changes) were identified that could potentially contribute to cancer development. Indeed, some genes can confer a selective growth advantage to cancerous cells when mutated, therefore, can promote tumorigenesis and/or cancer progression (= driver genes). These driver genes can be categorized to regulate three crucial cellular processes, namely cell fate, survival, and genome integrity [86, 87].
First indications that SAMHD1 might act as a driver gene were provided by a study from Schuh et al. tracking the clonal evolution in individual CLL patients during treatment [14]. In one patient, a somatic mutation in SAMHD1 was already detected in a founder subclone, suggesting it to be a potentially driving event in CLL [14]. Furthermore, subsequent whole-exome sequencing (WES) of CLL and germline DNA samples revealed that SAMHD1 was recurrently mutated in 2.5% (4/160) of CLL patients [44]. Although SAMDH1 mutations were found at lower frequencies compared to established CLL driver genes (like MYD88, TP53, or ATM), it was still classified as a potential driving cause in CLL [44]. All 20 potential driver genes are involved in seven specific signaling pathways (including DNA repair, cell cycle-control, and inflammatory pathways) known to be important in CLL [44]. As of today, SAMHD1’s involvement in several of these cellular processes could be shown (see section SAMHD1 function in this review), thereby directly linking SAMHD1 to these very same pathways involved in CLL development. However, SAMHD1’s role and impact as a potential driver gene might differ depending on CLL stage (for instance, before therapy and relapse after therapy). Using WES and deep resequencing, Amin et al. were able to examine paired samples (pre- and post-treatment) of 61 relapsed CLL patients, in order to uncover mutations in genes potentially driving CLL relapse [16]. First, SAMHD1 mutations were recurrently identified in 9.8% (6/61) of rCLL patients. In 53 paired DNA samples from patients before and after therapy, only mutations in TP53 and, to a lower extent, SAMHD1 were identified to be commonly enriched post-treatment: In 7.5% of rCLL patients (4/53), a marked enrichment of mutated SAMHD1 from already existing subclones could be detected at relapse [16]. These observations indicate that mutations in SAMHD1 are rather important to drive CLL relapse and/or impede CLL chemo-immunotherapy, than for early events during CLL onset/progression.
Additionally, SAMHD1 was only recently identified as a potential novel driver gene in MCL, with missense or deletion mutations in SAMHD1 being present in 10% of patients from the analyzed MCL cohort [88]. Of note, MCL can be subdivided into two molecular subgroups, namely conventional MCL (cMCL) and non-nodal MCL (nnMCL), which differ in their cellular origin, genetic features, and clinical outcome [24]. Compared to nnMCL, cMCL is characterized by a higher number of genetic aberrations (including structural variations and copy number alterations), although different mutation types occur at almost the same rate in both subtypes [88]. Nevertheless, mutations in SAMHD1 were only identified in cMCL patients; however, the present cohort comprised more cMCL (74%) compared to nnMCL (26%) cases, thereby not precluding that SAMHD1 mutations can also occur in nnMCL patients [88]. It will be interesting to explore the contribution of SAMHD1 mutations on the molecular level to MCL pathogenesis in future studies.
SAMHD1 seems not to be a highly mutated driver gene compared to known cancer drivers like TP53 (= tumor suppressor) or KRAS (= oncogene) [86, 89]. In some types of cancer, it could therefore be more difficult to identify SAMHD1 as a potential driver gene merely based on mutation frequency [86]. However, SAMHD1 was not recognized as a major driver gene in several studies using different methods which were not solely based on mutation frequency (e.g., [89, 90]).
Nevertheless, the pattern of cancer-related mutations that are found throughout the sequence of SAMHD1 (Fig. 4) together with the fact of its downregulation in several tumors (Fig. 3) rather indicates that SAMHD1 might act as a tumor suppressor. Therefore, future studies are needed to pinpoint which mutations in SAMHD1 might be true driver gene mutations (e.g., truncating, thereby inactivating, SAMHD1 or missense mutations influencing specific functions, like its dNTPase activity), not only passenger mutations that do not confer a selective growth advantage. Consequently, it is important to understand in which specific types of cancer and/or disease stages SAMHD1 mutations or expression level variations (e.g., through epigenetic changes) would promote malignant initiation and/or progression. With this knowledge, it will be possible to improve therapeutic efforts for individual malignancies or specific patients, as the genomic landscape of mutated (driver) genes can differ in each case appreciably.
Another interesting aspect has been discussed by Rentoft et al. [26]. Cancer-related, dNTPase-inactivating mutations in SAMHD1 might act in concert with other genetic defects (e.g., in DDR pathways like MMR) to promote a so-called mutator phenotype [26]. In this case, dNTP pool imbalances through SAMHD1 dysfunction in combination with defects in DNA repair/proofreading pathways could, potentially, lead to genomic instability, thereby favoring malignant transformation. SAMHD1 was proposed to rather act as a “mini driver” [26, 91], meaning that mutations in SAMHD1 might only slightly increase evolutionary fitness of tumor cells. However, in combination with mutations in other “mini-driver genes,” the effects of SAMHD1 mutation might add up and, ultimately, could lead to a growth advantage equivalent to major driver gene mutations [91], worthwhile to investigate. Therefore, future studies are needed to characterize SAMHD1-mutated cancer types and accompanying mutations in other, potential driver genes.
Outlook: SAMHD1 as a potential biomarker for treatment/clinical trials
As described, increasing evidence points towards a tumor suppressive role of SAMHD1 in different cancer types. However, several studies have shown by now that SAMHD1, due to its dNTPase activity, can also have a significant (negative) impact on the efficacy of nucleoside-based chemotherapies: For instance, different steps of AML therapy are often built around the nucleoside analog cytarabine (ara-C) which is converted intracellularly into the cytotoxic metabolite ara-C triphosphate (ara-CTP) [92]. SAMHD1 is able to degrade ara-CTP in vitro [78, 93, 94] and reduce its concentrations in cells like patient-derived AML blasts [78, 94], thereby posing a significant barrier to effective ara-C-based treatment. Indeed, SAMHD1 expression levels correlated with the effectiveness of ara-C therapy in different AML patient cohorts [78, 94]. High SAMHD1 expression correlated with a poorer response to ara-C-based AML induction [94], which was not observed in another study [78], as well as to consolidation therapy [78]. Therefore, SAMHD1 could be used as a marker to predict the outcome of ara-C-based regimens at different therapy stages [78, 94]. Additional studies are needed to further delineate how SAMHD1’s different functions (namely, being a barrier to ara-C-based therapies versus its potential tumor suppressive role) might influence each other, in order to predict the clinical response to ara-C of individual patients more precisely.
Additionally, SAMHD1 is able to hydrolyze several active triphosphate (TP) forms of nucleoside analogs used for anti-cancer therapies. Initial studies could show that SAMHD1 is able to degrade the TP form of clofarabine, which is used to treat pediatric ALL, at comparable rate to normal dNTPs [50, 95]. By comparing cytotoxicity of different nucleoside analogs in THP-1 cells and SAMHD1 knock-out THP1 cells, the resulting TP forms of vidarabine, nelarabine, fludarabine, trifluridine, and decitabine could be identified as potential SAMHD1 substrates [96], confirmed in structural studies [97]. Indeed, high SAMHD1 expression correlated with a poorer clinical response of AML patients to decitabine [98]. SAMHD1’s ability to degrade antimetabolites used in cancer therapy provides a rationale to directly alter the expression of SAMHD1, e.g., through Vpx-induced degradation [78, 94] or inhibit its activity using specific compounds to improve the efficacy of nucleoside-based chemotherapies.
Can SAMHD1 be regarded as a curse or cure for cancer? On the one hand, SAMHD1 appears to be a tumor suppressor, high expression correlates with a beneficial prognosis for many (but not all) cancers (Table 2), and mutations can have harmful effects (Table 1). However, SAMHD1 expression also interferes with nucleoside-based chemotherapeutics. One option would be to stratify patients based on their expression and mutation status to enable effective treatment. In conclusion, SAMHD1 can be both, curse and cure.
References
Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HCT, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW et al (2011) HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480(7377):379–382. https://doi.org/10.1038/nature10623
Franzolin E, Pontarin G, Rampazzo C, Miazzi C, Ferraro P, Palumbo E, Reichard P, Bianchi V (2013) The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc Natl Acad Sci USA 110(35):14272–14277. https://doi.org/10.1073/pnas.1312033110
Chen S, Bonifati S, Qin Z, St Gelais C, Kodigepalli KM, Barrett BS, Kim SH, Antonucci JM, Ladner KJ, Buzovetsky O et al (2018) SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-κB and interferon pathways. Proc Natl Acad Sci USA 115(16):E3798–E3807. https://doi.org/10.1073/pnas.1801213115
Daddacha W, Koyen AE, Bastien AJ, Head PE, Dhere VR, Nabeta GN, Connolly EC, Werner E, Madden MZ, Daly MB et al (2017) SAMHD1 promotes DNA end resection to facilitate DNA repair by homologous recombination. Cell Rep 20(8):1921–1935. https://doi.org/10.1016/j.celrep.2017.08.008
Coquel F, Silva M-J, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz A-L et al (2018) SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557(7703):57–61. https://doi.org/10.1038/s41586-018-0050-1
Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA et al (2009) Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41(7):829–832. https://doi.org/10.1038/ng.373
Coquel F, Neumayer C, Lin Y-L, Pasero P (2019) SAMHD1 and the innate immune response to cytosolic DNA during DNA replication. Curr Opin Immunol 56:24–30. https://doi.org/10.1016/j.coi.2018.09.017
Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kröger A, Nilsson JA et al (2015) DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42(2):332–343. https://doi.org/10.1016/j.immuni.2015.01.012
Lee EJ, Seo JH, Park J-H, Vo TTL, An S, Bae S-J, Le H, Lee HS, Wee H-J, Lee D et al (2017) SAMHD1 acetylation enhances its deoxynucleotide triphosphohydrolase activity and promotes cancer cell proliferation. Oncotarget 8(40):68517–68529. https://doi.org/10.18632/oncotarget.19704
Bonifati S, Daly MB, St Gelais C, Kim SH, Hollenbaugh JA, Shepard C, Kennedy EM, Kim D-H, Schinazi RF, Kim B et al (2016) SAMHD1 controls cell cycle status, apoptosis and HIV-1 infection in monocytic THP-1 cells. Virology 495:92–100. https://doi.org/10.1016/j.virol.2016.05.002
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
Zhang J, Bajari R, Andric D, Gerthoffert F, Lepsa A, Nahal-Bose H, Stein LD, Ferretti V (2019) The International Cancer Genome Consortium data portal. Nat Biotechnol 37(4):367–369. https://doi.org/10.1038/s41587-019-0055-9
Bosch F, Dalla-Favera R (2019) Chronic lymphocytic leukaemia: from genetics to treatment. Nat Rev Clin Oncol 16(11):684–701. https://doi.org/10.1038/s41571-019-0239-8
Schuh A, Becq J, Humphray S, Alexa A, Burns A, Clifford R, Feller SM, Grocock R, Henderson S, Khrebtukova I et al (2012) Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood 120(20):4191–4196. https://doi.org/10.1182/blood-2012-05-433540
Clifford R, Louis T, Robbe P, Ackroyd S, Burns A, Timbs AT, Wright Colopy G, Dreau H, Sigaux F, Judde JG et al (2014) SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood 123(7):1021–1031. https://doi.org/10.1182/blood-2013-04-490847
Amin NA, Seymour E, Saiya-Cork K, Parkin B, Shedden K, Malek SN (2016) A quantitative analysis of subclonal and clonal gene mutations before and after therapy in chronic lymphocytic leukemia. Clin Cancer Res 22(17):4525–4535. https://doi.org/10.1158/1078-0432.CCR-15-3103
Guièze R, Robbe P, Clifford R, de Guibert S, Pereira B, Timbs A, Dilhuydy M-S, Cabes M, Ysebaert L, Burns A et al (2015) Presence of multiple recurrent mutations confers poor trial outcome of relapsed/refractory CLL. Blood 126(18):2110–2117. https://doi.org/10.1182/blood-2015-05-647578
Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK (1999) Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 94(6):1848–1854
Burns A, Alsolami R, Becq J, Stamatopoulos B, Timbs A, Bruce D, Robbe P, Vavoulis D, Clifford R, Cabes M et al (2018) Whole-genome sequencing of chronic lymphocytic leukaemia reveals distinct differences in the mutational landscape between IgHVmut and IgHVunmut subgroups. Leukemia 32(2):332–342. https://doi.org/10.1038/leu.2017.177
Xin B, Jones S, Puffenberger EG, Hinze C, Bright A, Tan H, Zhou A, Wu G, Vargus-Adams J, Agamanolis D et al (2011) Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke. Proc Natl Acad Sci USA 108(13):5372–5377. https://doi.org/10.1073/pnas.1014265108
Merati M, Buethe DJ, Cooper KD, Honda KS, Wang H, Gerstenblith MR (2015) Aggressive CD8(+) epidermotropic cutaneous T-cell lymphoma associated with homozygous mutation in SAMHD1. JAAD Case Rep 1(4):227–229. https://doi.org/10.1016/j.jdcr.2015.05.003
Johansson P, Klein-Hitpass L, Choidas A, Habenberger P, Mahboubi B, Kim B, Bergmann A, Scholtysik R, Brauser M, Lollies A et al (2018) SAMHD1 is recurrently mutated in T-cell prolymphocytic leukemia. Blood Cancer J 8(1):11. https://doi.org/10.1038/s41408-017-0036-5
Roider T, Wang X, Hüttl K, Müller-Tidow C, Klapper W, Rosenwald A, Stewart JP, de Castro DG, Dreger P, Hermine O et al (2021) The impact of SAMHD1 expression and mutation status in mantle cell lymphoma: an analysis of the MCL Younger and Elderly trial. Int J Cancer 148(1):150–160. https://doi.org/10.1002/ijc.33202
Cortelazzo S, Ponzoni M, Ferreri AJM, Dreyling M (2020) Mantle cell lymphoma. Crit Rev Oncol Hematol 153:103038. https://doi.org/10.1016/j.critrevonc.2020.103038
Zhou L, Gu B, Shen X, Binshen O, Dong L, Zhou J, Yi H, Wang C (2021) B cell lymphoma with IRF4 rearrangement: a clinicopathological study of 13 cases. Pathol Int 71(3):183–190. https://doi.org/10.1111/pin.13067
Rentoft M, Lindell K, Tran P, Chabes AL, Buckland RJ, Watt DL, Marjavaara L, Nilsson AK, Melin B, Trygg J et al (2016) Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc Natl Acad Sci USA 113(17):4723–4728. https://doi.org/10.1073/pnas.1519128113
Powell RD, Holland PJ, Hollis T, Perrino FW (2011) Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J Biol Chem 286(51):43596–43600. https://doi.org/10.1074/jbc.C111.317628
Kunz BA, Kohalmi SE, Kunkel TA, Mathews CK, McIntosh EM, Reidy JA (1994) International Commission for Protection Against Environmental Mutagens and Carcinogens Deoxyribonucleoside triphosphate levels: a critical factor in the maintenance of genetic stability. Mutat Res 318(1):1–64. https://doi.org/10.1016/0165-1110(94)90006-x
Mathews CK (2015) Deoxyribonucleotide metabolism mutagenesis and cancer. Nat Rev Cancer 15(9):528–539. https://doi.org/10.1038/nrc3981
Schott K, Fuchs NV, Derua R, Mahboubi B, Schnellbächer E, Seifried J, Tondera C, Schmitz H, Shepard C, Brandariz-Nuñez A et al (2018) Dephosphorylation of the HIV-1 restriction factor SAMHD1 is mediated by PP2A-B55α holoenzymes during mitotic exit. Nat Commun 9(1):2227. https://doi.org/10.1038/s41467-018-04671-1
Yan J, Hao C, DeLucia M, Swanson S, Florens L, Washburn MP, Ahn J, Skowronski J (2015) CyclinA2-cyclin-dependent kinase regulates SAMHD1 protein phosphohydrolase domain. J Biol Chem 290(21):13279–13292. https://doi.org/10.1074/jbc.M115.646588
Tang C, Ji X, Wu L, Xiong Y (2015) Impaired dNTPase activity of SAMHD1 by phosphomimetic mutation of Thr-592. J Biol Chem 290(44):26352–26359. https://doi.org/10.1074/jbc.M115.677435
Aye Y, Li M, Long MJC, Weiss RS (2015) Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene 34(16):2011–2021. https://doi.org/10.1038/onc.2014.155
Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V et al (2018) Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173(2):291-304.e6. https://doi.org/10.1016/j.cell.2018.03.022
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2(5):401–404. https://doi.org/10.1158/2159-8290.CD-12-0095
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6(269):pl1. https://doi.org/10.1126/scisignal.2004088
White TE, Brandariz-Nuñez A, Martinez-Lopez A, Knowlton C, Lenzi G, Kim B, Ivanov D, Diaz-Griffero F (2017) A SAMHD1 mutation associated with Aicardi-Goutières syndrome uncouples the ability of SAMHD1 to restrict HIV-1 from its ability to downmodulate type I interferon in humans. Hum Mutat 38(6):658–668. https://doi.org/10.1002/humu.23201
Kretschmer S, Wolf C, König N, Staroske W, Guck J, Häusler M, Luksch H, Nguyen LA, Kim B, Alexopoulou D et al (2015) SAMHD1 prevents autoimmunity by maintaining genome stability. Ann Rheum Dis 74(3):e17. https://doi.org/10.1136/annrheumdis-2013-204845
Venkatesan RN, Bielas JH, Loeb LA (2006) Generation of mutator mutants during carcinogenesis. DNA Repair 5(3):294–302. https://doi.org/10.1016/j.dnarep.2005.10.012
Thientosapol ES, Bosnjak D, Durack T, Stevanovski I, van Geldermalsen M, Holst J, Jahan Z, Shepard C, Weninger W, Kim B et al (2018) SAMHD1 enhances immunoglobulin hypermutation by promoting transversion mutation. Proc Natl Acad Sci USA 115(19):4921–4926. https://doi.org/10.1073/pnas.1719771115
Husain A, Xu J, Fujii H, Nakata M, Kobayashi M, Wang J-Y, Rehwinkel J, Honjo T, Begum NA (2020) SAMHD1-mediated dNTP degradation is required for efficient DNA repair during antibody class switch recombination. EMBO J 39(15):e102931. https://doi.org/10.15252/embj.2019102931
Brandariz-Nuñez A, Valle-Casuso JC, White TE, Laguette N, Benkirane M, Brojatsch J, Diaz-Griffero F (2012) Role of SAMHD1 nuclear localization in restriction of HIV-1 and SIVmac. Retrovirology 9:49. https://doi.org/10.1186/1742-4690-9-49
Goncalves A, Karayel E, Rice GI, Bennett KL, Crow YJ, Superti-Furga G, Bürckstümmer T (2012) SAMHD1 is a nucleic-acid binding protein that is mislocalized due to aicardi-goutières syndrome-associated mutations. Hum Mutat 33(7):1116–1122. https://doi.org/10.1002/humu.22087
Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L et al (2013) Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152(4):714–726. https://doi.org/10.1016/j.cell.2013.01.019
Beloglazova N, Flick R, Tchigvintsev A, Brown G, Popovic A, Nocek B, Yakunin AF (2013) Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J Biol Chem 288(12):8101–8110. https://doi.org/10.1074/jbc.M112.431148
Rice GI, Forte GMA, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U et al (2013) Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1 RNASEH2A RNASEH2B RNASEH2C SAMHD1 and ADAR: a case-control study. Lancet Neurol 12(12):1159–1169. https://doi.org/10.1016/S1474-4422(13)70258-8
Yan J, Kaur S, DeLucia M, Hao C, Mehrens J, Wang C, Golczak M, Palczewski K, Gronenborn AM, Ahn J et al (2013) Tetramerization of SAMHD1 is required for biological activity and inhibition of HIV infection. J Biol Chem 288(15):10406–10417. https://doi.org/10.1074/jbc.M112.443796
Ji X, Tang C, Zhao Q, Wang W, Xiong Y (2014) Structural basis of cellular dNTP regulation by SAMHD1. Proc Natl Acad Sci USA 111(41):E4305–E4314. https://doi.org/10.1073/pnas.1412289111
Ji X, Wu Y, Yan J, Mehrens J, Yang H, DeLucia M, Hao C, Gronenborn AM, Skowronski J, Ahn J et al (2013) Mechanism of allosteric activation of SAMHD1 by dGTP. Nat Struct Mol Biol 20(11):1304–1309. https://doi.org/10.1038/nsmb.2692
Arnold LH, Groom HCT, Kunzelmann S, Schwefel D, Caswell SJ, Ordonez P, Mann MC, Rueschenbaum S, Goldstone DC, Pennell S et al (2015) Phospho-dependent regulation of SAMHD1 oligomerisation couples catalysis and restriction. PLoS Pathog 11(10):e1005194. https://doi.org/10.1371/journal.ppat.1005194
White TE, Brandariz-Nuñez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, Diaz-Griffero F (2013) The retroviral restriction ability of SAMHD1 but not its deoxynucleotide triphosphohydrolase activity is regulated by phosphorylation. Cell Host Microbe 13(4):441–451. https://doi.org/10.1016/j.chom.2013.03.005
Bhattacharya A, Wang Z, White T, Buffone C, Nguyen LA, Shepard CN, Kim B, Demeler B, Diaz-Griffero F, Ivanov DN (2016) Effects of T592 phosphomimetic mutations on tetramer stability and dNTPase activity of SAMHD1 can not explain the retroviral restriction defect. Sci Rep 6:31353. https://doi.org/10.1038/srep31353
Li Y, Kong J, Peng X, Hou W, Qin X, Yu X-F (2015) Structural insights into the high-efficiency catalytic mechanism of the sterile α-motif/histidine-aspartate domain-containing protein. J Biol Chem 290(49):29428–29437. https://doi.org/10.1074/jbc.M115.663658
Ryoo J, Choi J, Oh C, Kim S, Seo M, Kim S-Y, Seo D, Kim J, White TE, Brandariz-Nuñez A et al (2014) The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat Med 20(8):936–941. https://doi.org/10.1038/nm.3626
Seamon KJ, Bumpus NN, Stivers JT (2016) Single-stranded nucleic acids bind to the tetramer interface of SAMHD1 and prevent formation of the catalytic homotetramer. Biochemistry 55(44):6087–6099. https://doi.org/10.1021/acs.biochem.6b00986
Santos-Pereira JM, Aguilera A (2015) R loops: new modulators of genome dynamics and function. Nat Rev Genet 16(10):583–597. https://doi.org/10.1038/nrg3961
Park K, Ryoo J, Jeong H, Kim M, Lee S, Hwang S-Y, Ahn J, Kim D, Moon HC, Baek D et al (2021) Aicardi-Goutières syndrome-associated gene SAMHD1 preserves genome integrity by preventing R-loop formation at transcription-replication conflict regions. PLoS Genet 17(4):e1009523. https://doi.org/10.1371/journal.pgen.1009523
Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS et al (2012) Recurrent R-spondin fusions in colon cancer. Nature 488(7413):660–664. https://doi.org/10.1038/nature11282
Mao Z, Bozzella M, Seluanov A, Gorbunova V (2008) DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 7(18):2902–2906. https://doi.org/10.4161/cc.7.18.6679
Akimova E, Gassner FJ, Schubert M, Rebhandl S, Arzt C, Rauscher S, Tober V, Zaborsky N, Greil R, Geisberger R (2021) SAMHD1 restrains aberrant nucleotide insertions at repair junctions generated by DNA end joining. Nucleic Acids Res 49(5):2598–2608. https://doi.org/10.1093/nar/gkab051
Yang Y-G, Lindahl T, Barnes DE (2007) Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131(5):873–886. https://doi.org/10.1016/j.cell.2007.10.017
Li T, Chen ZJ (2018) The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215(5):1287–1299. https://doi.org/10.1084/jem.20180139
Franzolin E, Coletta S, Ferraro P, Pontarin G, D′Aronco G, Stevanoni M, Palumbo E, Cagnin S, Bertoldi L, Feltrin E, et al (2020) SAMHD1-deficient fibroblasts from Aicardi-Goutières Syndrome patients can escape senescence and accumulate mutations. FASEB J 34(1):631–647. https://doi.org/10.1096/fj.201902508R
Gubernatorova EO, Polinova AI, Petropavlovskiy MM, Namakanova OA, Medvedovskaya AD, Zvartsev RV, Telegin GB, Drutskaya MS, Nedospasov SA (2021) Dual role of TNF and LTα in carcinogenesis as implicated by studies in mice. Cancers 13(8):1775. https://doi.org/10.3390/cancers13081775
Balkwill F (2006) TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev 25(3):409–416. https://doi.org/10.1007/s10555-006-9005-3
Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob L, Patwa R, Shah H, Xu K et al (2016) Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533(7604):493–498. https://doi.org/10.1038/nature18268
Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN (2014) Inflammation-driven carcinogenesis is mediated through STING. Nat Commun 5:5166. https://doi.org/10.1038/ncomms6166
Qin Z, Bonifati S, St Gelais C, Li T-W, Kim S-H, Antonucci JM, Mahboubi B, Yount JS, Xiong Y, Kim B et al (2020) The dNTPase activity of SAMHD1 is important for its suppression of innate immune responses in differentiated monocytic cells. J Biol Chem 295(6):1575–1586. https://doi.org/10.1074/jbc.RA119.010360
Baylin SB, Jones PA (2011) A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11(10):726–734. https://doi.org/10.1038/nrc3130
de Silva S, Wang F, Hake TS, Porcu P, Wong HK, Wu L (2014) Downregulation of SAMHD1 expression correlates with promoter DNA methylation in Sézary syndrome patients. J Invest Dermatol 134(2):562–565. https://doi.org/10.1038/jid.2013.311
Kiel MJ, Sahasrabuddhe AA, Rolland DCM, Velusamy T, Chung F, Schaller M, Bailey NG, Betz BL, Miranda RN, Porcu P et al (2015) Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sézary syndrome. Nat Commun 6:8470. https://doi.org/10.1038/ncomms9470
de Silva S, Hoy H, Hake TS, Wong HK, Porcu P, Wu L (2013) Promoter methylation regulates SAMHD1 gene expression in human CD4+ T cells. J Biol Chem 288(13):9284–9292. https://doi.org/10.1074/jbc.M112.447201
Kohnken R, Kodigepalli KM, Mishra A, Porcu P, Wu L (2017) MicroRNA-181 contributes to downregulation of SAMHD1 expression in CD4+ T-cells derived from Sèzary syndrome patients. Leukemia Res 52:58–66. https://doi.org/10.1016/j.leukres.2016.11.010
Kodigepalli KM, Li M, Liu S-L, Wu L (2017) Exogenous expression of SAMHD1 inhibits proliferation and induces apoptosis in cutaneous T-cell lymphoma-derived HuT78 cells. Cell Cycle 16(2):179–188. https://doi.org/10.1080/15384101.2016.1261226
Hunger SP, Mullighan CG (2015) Acute lymphoblastic leukemia in children. N Engl J Med 373(16):1541–1552. https://doi.org/10.1056/NEJMra1400972
Rothenburger T, McLaughlin K-M, Herold T, Schneider C, Oellerich T, Rothweiler F, Feber A, Fenton TR, Wass MN, Keppler OT et al (2020) SAMHD1 is a key regulator of the lineage-specific response of acute lymphoblastic leukaemias to nelarabine. Commun Biol 3(1):324. https://doi.org/10.1038/s42003-020-1052-8
DiNardo CD (2016) Cortes JE (2016) Mutations in AML: prognostic and therapeutic implications. Hematology 1:348–355. https://doi.org/10.1182/asheducation-2016.1.348
Herold N, Rudd SG, Ljungblad L, Sanjiv K, Myrberg IH, Paulin CBJ, Heshmati Y, Hagenkort A, Kutzner J, Page BDG et al (2017) Targeting SAMHD1 with the Vpx protein to improve cytarabine therapy for hematological malignancies. Nat Med 23(2):256–263. https://doi.org/10.1038/nm.4265
Jiang H, Li C, Liu Z, Shengjing H (2020) Expression and relationship of SAMHD1 with other apoptotic and autophagic genes in acute myeloid leukemia patients. Acta Haematol 143(1):51–59. https://doi.org/10.1159/000500822
Xagoraris I, Vassilakopoulos TP, Drakos E, Angelopoulou MK, Panitsas F, Herold N, Medeiros LJ, Giakoumis X, Pangalis GA, Rassidakis GZ (2021) Expression of the novel tumour suppressor sterile alpha motif and HD domain-containing protein 1 is an independent adverse prognostic factor in classical Hodgkin lymphoma. Br J Haematol 193(3):488–496. https://doi.org/10.1111/bjh.17352
Wang J, Lu F, Shen X-Y, Wu Y, Zhao L (2014) SAMHD1 is down regulated in lung cancer by methylation and inhibits tumor cell proliferation. Biochem Biophys Res Commun 455(3–4):229–233. https://doi.org/10.1016/j.bbrc.2014.10.153
Wu Y, Niu Y, Wu Y, Chen X, Shen X, Gao W (2021) SAMHD1 can suppress lung adenocarcinoma progression through the negative regulation of STING. J Thorac Dis 13(1):189–201. https://doi.org/10.21037/jtd-20-1889
Leonardi GC, Falzone L, Salemi R, Zanghì A, Spandidos DA, Mccubrey JA, Candido S, Libra M (2018) Cutaneous melanoma: from pathogenesis to therapy (Review). Int J Oncol 52(4):1071–1080. https://doi.org/10.3892/ijo.2018.4287
Chen W, Cheng P, Jiang J, Ren Y, Wu D, Xue D (2020) Epigenomic and genomic analysis of transcriptome modulation in skin cutaneous melanoma. Aging 12(13):12703–12725. https://doi.org/10.18632/aging.103115
Herold N, Rudd SG, Sanjiv K, Kutzner J, Myrberg IH, Paulin CBJ, Olsen TK, Helleday T, Henter J-I, Schaller T (2017) With me or against me: tumor suppressor and drug resistance activities of SAMHD1. Exp Hematol 52:32–39. https://doi.org/10.1016/j.exphem.2017.05.001
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (2013) Cancer genome landscapes Sci 339(6127):1546–1558. https://doi.org/10.1126/science.1235122
Pon JR, Marra MA (2015) Driver and passenger mutations in cancer. Ann Rev Pathol 10:25–50. https://doi.org/10.1146/annurev-pathol-012414-040312
Nadeu F, Martin-Garcia D, Clot G, Díaz-Navarro A, Duran-Ferrer M, Navarro A, Vilarrasa-Blasi R, Kulis M, Royo R, Gutiérrez-Abril J et al (2020) Genomic and epigenomic insights into the origin pathogenesis and clinical behavior of mantle cell lymphoma subtypes. Blood 136(12):1419–1432. https://doi.org/10.1182/blood.2020005289
Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, Mularoni L, Pich O, Bonet J, Kranas H et al (2020) A compendium of mutational cancer driver genes. Nat Rev Cancer 20(10):555–572. https://doi.org/10.1038/s41568-020-0290-x
Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B et al (2018) Comprehensive characterization of cancer driver genes and mutations. Cell 174(4):1034–1035. https://doi.org/10.1016/j.cell.2018.07.034
Castro-Giner F, Ratcliffe P, Tomlinson I (2015) The mini-driver model of polygenic cancer evolution. Nat Rev Cancer 15(11):680–685. https://doi.org/10.1038/nrc3999
Döhner H, Weisdorf DJ, Bloomfield CD (2015) Acute myeloid leukemia. N Engl J Med 373(12):1136–1152. https://doi.org/10.1056/NEJMra1406184
Hollenbaugh JA, Shelton J, Tao S, Amiralaei S, Liu P, Lu X, Goetze RW, Zhou L, Nettles JH, Schinazi RF et al (2017) Substrates and inhibitors of SAMHD1. PLoS ONE 12(1):e0169052. https://doi.org/10.1371/journal.pone.0169052
Schneider C, Oellerich T, Baldauf H-M, Schwarz S-M, Thomas D, Flick R, Bohnenberger H, Kaderali L, Stegmann L, Cremer A et al (2017) SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat Med 23(2):250–255. https://doi.org/10.1038/nm.4255
Ordonez P, Kunzelmann S, Groom HCT, Yap MW, Weising S, Meier C, Bishop KN, Taylor IA, Stoye JP (2017) SAMHD1 enhances nucleoside-analogue efficacy against HIV-1 in myeloid cells. Sci Rep 7:42824. https://doi.org/10.1038/srep42824
Herold N, Rudd SG, Sanjiv K, Kutzner J, Bladh J, Paulin CBJ, Helleday T, Henter J-I, Schaller T (2017) SAMHD1 protects cancer cells from various nucleoside-based antimetabolites. Cell Cycle 16(11):1029–1038. https://doi.org/10.1080/15384101.2017.1314407
Knecht KM, Buzovetsky O, Schneider C, Thomas D, Srikanth V, Kaderali L, Tofoleanu F, Reiss K, Ferreirós N, Geisslinger G et al (2018) The structural basis for cancer drug interactions with the catalytic and allosteric sites of SAMHD1. Proc Natl Acad Sci USA 115(43):E10022–E10031. https://doi.org/10.1073/pnas.1805593115
Oellerich T, Schneider C, Thomas D, Knecht KM, Buzovetsky O, Kaderali L, Schliemann C, Bohnenberger H, Angenendt L, Hartmann W et al (2019) Selective inactivation of hypomethylating agents by SAMHD1 provides a rationale for therapeutic stratification in AML. Nat Commun 10(1):3475. https://doi.org/10.1038/s41467-019-11413-4
Zhao K, Du J, Han X, Goodier JL, Li P, Zhou X, Wei W, Evans SL, Li L, Zhang W et al (2013) Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutières syndrome-related SAMHD1. Cell Rep 4(6):1108–1115. https://doi.org/10.1016/j.celrep.2013.08.019
Ahn J (2016) Functional organization of human SAMHD1 and mechanisms of HIV-1 restriction. Biol Chem 397(4):373–379. https://doi.org/10.1515/hsz-2015-0260
Zhu C, Gao W, Zhao K, Qin X, Zhang Y, Peng X, Zhang L, Dong Y, Zhang W, Li P et al (2013) Structural insight into dGTP-dependent activation of tetrameric SAMHD1 deoxynucleoside triphosphate triphosphohydrolase. Nat Commun 4:2722. https://doi.org/10.1038/ncomms3722
Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L et al (2011) SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365(26):2497–2506. https://doi.org/10.1056/NEJMoa1109016
Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M, Böttcher S et al (2015) Mutations driving CLL and their evolution in progression and relapse. Nature 526(7574):525–530. https://doi.org/10.1038/nature15395
Seamon KJ, Sun Z, Shlyakhtenko LS, Lyubchenko YL, Stivers JT (2015) SAMHD1 is a single-stranded nucleic acid binding protein with no active site-associated nuclease activity. Nucleic Acids Res 43(13):6486–6499. https://doi.org/10.1093/nar/gkv633
Walker BA, Wardell CP, Melchor L, Hulkki S, Potter NE, Johnson DC, Fenwick K, Kozarewa I, Gonzalez D, Lord CJ et al (2012) Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood 120(5):1077–1086. https://doi.org/10.1182/blood-2012-03-412981
Jay JJ, Brouwer C (2016) Lollipops in the clinic: information dense mutation plots for precision medicine. PLoS ONE 11(8):e0160519. https://doi.org/10.1371/journal.pone.0160519
Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F et al (2017) A pathology atlas of the human cancer transcriptome. Science (New York, N.Y.) 357(6352). https://doi.org/10.1126/science.aan2507
Funding
Open Access funding enabled and organized by Projekt DEAL. This project was supported by the German Research Foundation (DFG), Collaborative Research Center 1292, project TP09, TP05, TP06, and TP04.
Author information
Authors and Affiliations
Contributions
AB and LC provided data analysis, figures, tables, and related figure text; KS, CM, and RK drafted the review and provided figures and tables; MHHS, KR, and MM critically reviewed the content and edited the text. RK conceived, edited, and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kerstin Schott, Catharina Majer, and Alla Bulashevska contributed equally to this work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schott, K., Majer, C., Bulashevska, A. et al. SAMHD1 in cancer: curse or cure?. J Mol Med 100, 351–372 (2022). https://doi.org/10.1007/s00109-021-02131-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-021-02131-w