
Abstract
The clinical manifestations of COVID-19 are mainly respiratory symptoms, but some patients present with cardiovascular system disease such as palpitations and shortness of breath as the first or secondary symptoms. In this paper, we describe the characteristics of SARS-CoV‑2 and its functional receptor angiotensin-converting enzyme 2 (ACE2). Furthermore, we explore the impact of virus-induced myocardial damage, decreased ACE2 activity, immune imbalance, hypoxemia, and heart damage caused by antiviral drugs.
Zusammenfassung
Das klinische Bild einer Infektion mit COVID-19 zeigt sich hauptsächlich in Atemwegssymptomen, einige Patienten weisen jedoch eine Beteiligung des kardiovaskulären Systems auf – mit Palpitationen und Kurzatmigkeit als Erstsymptomen oder als sekundär auftretenden Symptomen. In der vorliegenden Arbeit werden die Charakteristika von SARS-CoV‑2 und seinem funktionalen Rezeptor Angiotensin-Converting-Enzym 2 (ACE2) dargestellt. Darüber hinaus wird der Einfluss von virusinduzierten Myokardläsionen, erniedrigter ACE2-Aktivität, eines Ungleichgewichts im Immunsystem, von Hypoxämie und einer durch antivirale Medikamente verursachten Herzinsuffizienz untersucht.
Similar content being viewed by others

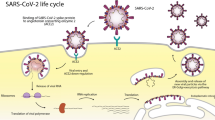
The clinical manifestations of COVID-19 are mainly respiratory symptoms, but some patients present with cardiovascular system damage such as palpitations and shortness of breath as the first or secondary symptoms. In addition, some people with basic cardiovascular disease (CVD) may have an increased risk of death. Therefore, it is important to understand the potential mechanisms of SARS-CoV‑2 damage to the cardiovascular system in order to provide timely and effective treatment and reduce mortality in these patients.
Virological characteristics of SARS-CoV-2
Vertebral coronaviruses are widespread in humans and several other animals, causing a variety of acute and chronic diseases. According to the serotype and genome characteristics, the coronavirus subfamily is divided into four genera: α, β, γ, and δ [1]. The crown-like appearance of this virus under the electron microscope lent it the name “coronavirus”. Among the seven coronavirus subtypes that can infect humans, the α genus coronavirus has low pathogenicity, with asymptomatic or self-limiting upper respiratory tract infection symptoms after infection, and the β genus coronavirus causes lower respiratory tract diseases such as pneumonia or bronchitis. Patients with cardiopulmonary disease, immunocompromised individuals, infants, and older people may experience serious illness and death [2]. SARS-CoV‑2 is a linear single-stranded RNA enveloped virus belonging to the β‑CoV category, closely related to SARS-CoV virus, and has the ability of rapid mutation and recombination [3]. The virus particle is enveloped in a fatty membrane, and the spike protein is on the surface of the envelope, which, as one of the major antigenic proteins of the virus, can bind to the angiotensin-converting enzyme 2 (ACE2) receptor between the envelope and the host cell membrane to help the virus enter the host cell [4].
ACE2, a functional receptor of SARS-CoV-2
Angiotensin-converting enzyme 2 is a membrane-bound aminopeptidase that is highly expressed in alveolar cells and plays an important role in the cardiovascular and immune systems. It is involved in the development of heart function, hypertension, and diabetes. In addition, ACE2 has been identified as a functional receptor for coronaviruses (including SARS-CoV and SARS-CoV‑2; [5]). SARS-CoV‑2 infection is caused by the viral membrane protein binding to the ACE2 receptor on the surface of the host cell and fusing with the host cell membrane, entering the cell through receptor-mediated endocytosis in a manner similar to that of human immunodeficiency virus (HIV; [6]). It causes endocytosis of the ligand/receptor complex, induces fusion of the virus and the host cell, and subsequently, ACE2 is degraded within the cell.
Since the outbreak of SARS-CoV in 2002, extensive structural analysis from the atomic level has revealed that ACE2 can regulate the transmission of SARS-CoV across species and between people [7]. Surface plasmon resonance technology confirmed that the binding force between membrane protein and ACE2 of SARS-CoV‑2 was 10–20 times higher than that of SARS-CoV, suggesting that SARS-CoV‑2 was more infectious than SARS-CoV at the genetic level [8]. This is also the reason for the spread of SARS-CoV‑2 worldwide, and consistent with epidemiological results, people of all ages may be infected. ACE2 is highly expressed in the heart and lungs, and SARS-CoV‑2 mainly invades alveolar epithelial cells, leading to respiratory symptoms. These symptoms are more severe in patients with CVD, which may be related to increased ACE2 secretion in these patients compared with healthy individuals. Because ACE2 has a lung protective effect, when healthy people are infected with SARS-CoV‑2, membrane proteins bind to ACE2 of the alveolar epithelial cells allowing the virus to disrupt the ACE2 lung protection pathway, resulting in viral pathogenesis [9].
The renin–angiotensin system (RAS) is a key factor that controls the cardiovascular system and the balance of water and electrolytes, affecting the body’s organs and functions [10]. Both ACE2 and ACE have a balance effect on the RAS in the steady-state control of the cardiovascular and renal systems and in regulating the volume of extracellular fluid. There are two main pathways: one is the classic RAS pathway, which is the ACE/AngII/AT1R pathway, and the second axis is ACE2/ang1-7/Mas and ACE2/ang1-9/AT2R, which balance each other. ACE2 activity leads to AngII/AT1R axis activation, and plays an important role in promoting the development of CVD. This strongly suggests that ACE2 is an important regulator of cardiac function in vivo [11, 12]. SARS-CoV‑2 uses ACE2 as a functional receptor to enter human cells. Therefore, ACE2-related pathways may play a role in COVID-19 cardiovascular damage.
Effect and mechanism of SARS-CoV2 on cardiovascular system
In addition to typical respiratory manifestations, a proportion of COVID-19 patients have clinical manifestations of cardiac involvement. Among the confirmed cases of SARS-CoV‑2 infection reported by the National Health Commission of China (NHC), some patients were treated first for cardiovascular symptoms. Among those who died of COVID-19, 11.8% of patients without basic CVD had significant heart damage, increased cardiac troponin I levels, or cardiac arrest during hospitalization, and the percentage of deaths from COVID-19 infection with hypertension, diabetes, and coronary heart disease was much higher than other populations [13].
Acute myocardial injury due to SARS-CoV-2 infection
Previous studies in other coronavirus genera (MERS-CoV) confirmed the signs of acute myocarditis by cardiac magnetic resonance imaging [14]. Studies have confirmed that in 35% (7/20) of autopsy heart samples from patients who died of SARS crisis during the SARS outbreaks, RNA of the SARS-CoV virus was detected [15] In pathological examinations of SARS patients, it was found that the heart showed cardiomyocyte degeneration and atrophy. Nucleic acid in situ hybridization showed coronavirus-positive cardiomyocytes in the cardiac conduction system. In autopsy studies of SARS deaths, virus-like particles were found in the cytoplasm of myocardial cells. A small number of myocardial cells were degenerated and atrophied, suggesting that the virus causes direct damage to the myocardium [16]. Homology modeling showed that SARS-CoV‑2 and SARS-CoV have similar receptor binding domain structures, the main proteases are highly conserved, and the overall identity is 96%, but the pathogenicity and spread of SARS-CoV‑2 are higher than those of SARS-CoV [17]. Therefore, the myocardial damage caused by SARS-CoV‑2 infection undoubtedly increases the difficulty and complexity of treatment for patients.
Activity of ACE2 is decreased by SARS CoV-2 infection
The mechanism of acute myocardial injury caused by SARS-CoV‑2 infection may be related to ACE2. ACE2 is widely expressed in the pulmonary and cardiovascular systems, and thus ACE2-related signaling pathways may also play a role in heart injury. The expression of ACE2 was significantly reduced in mice with pulmonary infection of human SARS-CoV, resulting in ACE2-dependent myocardial infection and confirming that ACE2 plays a key role in mediating SARS-CoV infection in the heart [18]. ACE2 has been identified as an important RAS regulator that alleviates AngII- and ATR-mediated deleterious effects. With the increase or decrease of ACE2, the content of Ang(1-7) and Ang(1-9), which have a protective effect on the cardiovascular system, will also change, the balance state between the ACE/AngII/AT1R axis and the ACE2/Ang1-7/Mas and ACE2/Ang1-9/AT2R axis is broken, and the AngII/AT1R axis is activated accordingly [19, 20]. Plasma AngII levels in COVID-19 patients are clearly higher than those of healthy individuals, and is closely related to the viral load and the degree of lung injury [21]; ACE and AngII are poor prognostic factors of severe pneumonia. The results of animal experiments show that RAS inhibitor can effectively relieve the symptoms of acute severe pneumonia and respiratory failure. Novel coronavirus binding with ACE2 leads to a sharp decrease in ACE2, inhibition of the ACE2/Ang (1-7)/Mas receptor pathway, and an imbalance of the RAS system, which may induce acute myocardial injury, acute coronary syndrome, arrhythmia, heart failure, etc., leading to an increased case fatality rate in COVID-19 patients [22].
Immune imbalance and cytokine storm
Cytokine storms themselves have been found to be a major cause of morbidity and mortality from influenza viruses and other acute and severe respiratory infections, rather than being a concomitant phenomenon [23]. The SARS-CoV virus was detected in autopsy heart samples from patients who died of SARS infection, and macrophage-specific staining showed a significant increase in cardiac macrophage infiltration and evidence of myocardial injury [18]. Wong et al. proposed that the mechanisms of myocardial injury in patients diagnosed with SARS include cytokine storms triggered by an unbalanced response of type 1 and type 2 helper T cells [24]. On the basis of clinical data, it was found that in 41 patients with laboratory-confirmed SARS-CoV-2 infection, plasma interleukin IL-1B, IL‑1RA, IL‑7, IL‑8, IL‑9, IL-10, FGF, G-CSF, GM-CSF, IFN-γ, IP-10, MCP-1, MIP-1A, MIP-1B, PDGF, TNF-α, and VEGF are higher than normal. Moreover, serum cytokine concentrations of patients requiring admission to the intensive care unit are higher than those of patients on general wards, indicating that the degree of immune imbalance is related to the severity of the disease [25].
The autopsy of the first patient who died of severe SARS-CoV‑2 infection showed that the overactivation of T cells led to severe immune injury, and there was a small amount of inflammatory infiltration of monocytes in the myocardial interstitium [26]. Initially, SARS-CoV‑2 mainly infects the lower respiratory tract and binds to ACE2 on alveolar epithelial cells and on the heart. SARS-CoV‑2 virus is a powerful inducer of inflammatory cytokines [27]. Therefore, the immune imbalance caused by SARS-CoV2 infection and the cytokine storm may be one of the mechanisms of myocardial injury. Due to the systemic inflammatory response and immune system diseases during disease progression, the incidence of cardiovascular symptoms is higher.
Hypoxemia, imbalance of acid–base balance, and electrolyte disorder
The main clinical manifestation of COVID-19 is acute respiratory disease, which may cause acute respiratory distress syndrome (ARDS), presenting as ground-glass shadow and hypoxemia on chest imaging [28]. Chronic and severe intermittent hypoxia can lead to persistent and maladaptive chemical-mediated activation of the sympathetic nervous system [29], and also lead to persistent hypertension [29], RAS activation [30], increased oxidative stress [31], systemic inflammation, activation of NF-B-mediated inflammatory pathways [32], and an increase in the inflammatory cytokines TNF‑α and IL‑6 as well as increased expression of C‑reactive protein (CRP; [33]). Inflammation may cause endothelial dysfunction and injury and promote the formation of atherosclerosis [34]. Anoxia can also lead to inactivation of nitric oxide, acidosis, electrolyte disturbance, and the generation of oxygen free radicals, which all aggravate myocardial injury and combine to cause further damage to the cardiovascular system [35]. Half of the patients with COVID-19 infection developed dyspnea after a week, and in severe cases, they developed ARDS, septic shock, refractory metabolic acidosis, and coagulation dysfunction [28].
Heart damage caused by antiviral drugs
Antiviral drugs are the most important drugs for the treatment of COVID-19. However, many antiviral drugs can cause cardiac dysfunction, arrhythmias, or other CVD. Lopinavir and ritonavir are recommended as first-line antiviral drugs in the “National protocol for the diagnosis and treatment of COVID-19”. These two drugs inhibit the replication of SARS-CoV‑2 virus RNA, may lead to prolonged QT and PR intervals, and may accelerate the progression of coronary heart disease [36]. When combined with anticoagulant drugs such as ribavirin and warfarin, the dose-effect of warfarin was clinically significantly reduced leading to a risk of bleeding [37]. Remdesivir has been approved by the FDA for emergency use in patients with suspected or laboratory-confirmed COVID-19 [38]. A previous evaluation of the drug during the Ebola outbreak found that one patient (out of a total of 175) experienced hypotension and subsequent cardiac arrest after receiving the loaded dose [39]. As an antimalarial drug, chloroquine has been proved to inhibit the activity of SARS-CoV‑2 in vitro and has been widely used in clinical practice. However, chloroquine can cause cardiac arrhythmia, and the most serious adverse reaction of chloroquine phosphate is cardiac arrest [40]. Abidor is a non-nucleoside broad-spectrum antiviral drug, which has an anti-SARS coronavirus effect in vitro but has potential side effects on the heart. Some healthy subjects developed bradycardia 3 h after taking the drug [41]. Methylprednisolone, a steroid currently used to treat severe cases of COVID-19 with ARDS, is known to cause fluid retention, electrolyte disturbances, and hypertension [42]. Therefore, the risk of cardiotoxicity must be closely monitored during the treatment of COVID-19, especially with antiviral drugs.
Conclusion
Novel coronavirus (2019-nCoV) mainly affects alveolar epithelial cells through the angiotensin-converting enzyme 2 (ACE2) receptor and causes pulmonary inflammation resulting in respiratory symptoms. However, novel coronavirus also invades the myocardium directly through ACE2 and causes a series of pathophysiological changes. Therefore, patients with pre-existing cardiovascular diseases should be given high priority in clinical practice to avoid the development of severe or critical coronavirus disease.
References
Wong ACP et al (2019) Global epidemiology of bat coronaviruses. Viruses 11(2):174–196
Zhou P et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273
Velavan TP, Meyer CG (2020) The COVID-19 epidemic. Trop Med Int Health 25(3):278–280
Perlman S, Netland J (2009) Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7(6):439–450
Turner AJ et al (2004) ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol Sci 25(6):291–294
Wang H et al (2008) SARS coronavirus entry into host cells through a novel clathrin-and caveolae-independent endocytic pathway. Cell Res 18(2):290–301
Wan Y et al (2020) Receptor recognition by novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS. J Virol. https://doi.org/10.1128/JVI.00127-20
Wrapp D et al (2020) Cryo-EMstruc-tureofthe2019-nCoV spike in the prefusion conformation. Science 367(6483):1260–1263
Zhang H et al (2020) Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV‑2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 3:1–5
Souza Santos RA et al (2018) The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1-7). Physiol Rev 98(1):505–553
Miller AJ, Arnold AC (2019) The renin-angiotensin system in cardiovascular autonomic control: recent developments and clinical implications. Clin Auton Res 29(2):231–243
Oliveira Andrade JM et al (2017) The angiotensin converting enzyme 2 (ACE2), gut microbiota, and cardiovascular health. Protein Pept Lett 24(9):827–832
Wang D et al (2020) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323(11):1061–1069
Alhogbani T (2016) Acute myocarditis associated with novel Middle East respiratory syndrome coronavirus. Ann Saudi Med 36(1):78–80
Grant PR et al (2003) Detection of SARS coronavirus in plasma by real-time RT-PCR. N Engl J Med 349(25):2468–2469
Zhao J et al (2003) Clinicopathology and pathogenesis of severe acute respiratory syndrome. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi 17(3):217–223
Khan S et al (2020) The emergence of a novel coronavirus (SARS-coV-2), their biology and therapeutic options. J Clin Microbiol 3:187–120
Oudit GY et al (2009) SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest 39(7):618–625
Simões E Silva AC, Martins Teixeira M (2016) ACE inhibition, ACE2 and angiotensin-(1-7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol Res 107:154–162
Mendoza-Torres E et al (2015) ACE2 and vasoactive peptides: novel players in cardiovascular/renal remodeling and hypertension. Ther Adv Cardiovasc Dis 9(4):217–237
Liu Y et al (2020) Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung in jury. Sci China Life Sci 63(3):364–374
Sun ML et al (2020) Inhibitors of RAS might be a good choice for the therapy of COVID-19 pneumonia. Zhonghua Jie He He Hu Xi Za Zhi 43(3):219–222
Walsh KB et al (2011) Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci U S A 108(29):12018–12023
Wong CK et al (2004) Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol 136(1):95–103
Huang C et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223):497–506
Xu Z et al (2020) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 18:S2213–2600
Jiang F et al (2020) Review of the clinical characteristics of coronavirus disease 2019 (COVID-19). J Gen Intern Med 35(5):1545–1549
Deng SQ, Peng HJ (2020) Characteristics of and public health responses to the coronavirus disease 2019 outbreak in China. J Clin Med 9(2):575–675
Prabhakar NR et al (2005) Cardiovascular alterations by chronic intermittent hypoxia: importance of carotid body chemoreflexes. Clin Exp Pharmacol Physiol 32(5–6):447–449
Foster GE et al (2010) Intermittent hypoxia increases arterial blood pressure in humans through a renin-angiotensin system-dependent mechanism. Hypertension 56:369–377
Troncoso Brindeiro CM et al (2007) Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. Am J Physiol Heart Circ Physiol 293:H2971–H2976
Ryan S et al (2006) Predictors of elevated nuclear factor-B-dependent genes in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 174:824–830
Saito M et al (2003) Relations of plasma high-sensitivity C‑reactive protein to traditional cardiovascular risk factors. Atherosclerosis 167(1):73–79
Wolf D, Ley K (2019) Immunity and inflammation in atherosclerosis. Circ Res 124(2):315–327
Beaudin AE et al (2017) Impact of obstructive sleep apnoea and intermittent hypoxia on cardiovascular and cerebrovascular regulation. Exp Physiol 102(7):743–763
Park SY et al (2019) Post-exposure prophylaxis for middle east respiratory syndrome in Healthcare workers. J Hosp Infect 101(1):42–46
DeCarolis DD et al (2017) Evaluation of a potential interaction between new regimens to treat hepatitis C and warfarin: twelve-week post-treatment follow-up. Ann Pharmacother 51(5):439–440
Hashemian SM et al (2020) A review on remdesivir: a possible promising agent for the treatment of COVID-19. Drug Des Devel Ther 14:3215–3222
Mulangu S et al (2019) A randomized, controlled trial of ebola virus disease therapeutics. N Engl J Med 381:2293–2303
Gao J et al (2020) Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci Trends 14(1):72–73
Zhong Q et al (2009) Antiviral activity of arbidol against coxsackie virus B5 in vitro and in vivo. Arch Virol 154(4):601–607
Liu Y et al (2020) Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci 63:364–374
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
M. Li, S. Chen, X. Xiang, Q. Wang and X. Liu declare that they have no competing interests.
For this article no studies with human participants or animals were performed by any of the authors. All studies performed were in accordance with the ethical standards indicated in each case.
Additional information
Li Mingzhe, Chen Siyang, Xiang Xiaochen contributed equally to this paper.
Rights and permissions
About this article

Cite this article
Li, M., Chen, S., Xiang, X. et al. Effects of SARS-CoV-2 and its functional receptor ACE2 on the cardiovascular system. Herz 45, 659–662 (2020). https://doi.org/10.1007/s00059-020-04989-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00059-020-04989-x