
Abstract
Phosphoinositides help steer membrane trafficking routes within eukaryotic cells. In polarized exocytosis, which targets vesicular cargo to sites of polarized growth at the plasma membrane (PM), the two phosphoinositides phosphatidylinositol 4-phosphate (PI4P) and its derivative phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) pave the pathway for vesicle transport from the Golgi to the PM. PI4P is a critical regulator of mechanisms that shape late Golgi membranes for vesicle biogenesis and release. Although enriched in vesicle membranes, PI4P is inexplicably removed from post-Golgi vesicles during their transit to the PM, which drives subsequent steps in exocytosis. At the PM, PI(4,5)P2 recruits effectors that establish polarized membrane sites for targeting the vesicular delivery of secretory cargo. The budding yeast Saccharomyces cerevisiae provides an elegant model to unravel the complexities of phosphoinositide regulation during polarized exocytosis. Here, we review how PI4P and PI(4,5)P2 promote yeast vesicle biogenesis, exocyst complex assembly and vesicle docking at polarized cortical sites, and suggest how these steps might impact related mechanisms of human disease.
Similar content being viewed by others


Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Introduction
The transport pathway for exocytosis, from post-Golgi vesicle biogenesis to vesicle targeting sites on the plasma membrane (PM), is promoted by phosphoinositide lipid gradients. Phosphoinositides are phosphorylated derivatives of phosphatidylinositol (PI) that are recognized by specific domains on proteins for their recruitment at membranes. Although distinct sets of phosphoinositides demarcate different intracellular membranes, the most relevant phosphoinositides involved in exocytosis are phosphatidylinositol 4-phosphate (PI4P) and its derivative phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2]. In budding yeast, PI4P is mainly enriched in the Golgi and nascent vesicles that bud off of the trans-Golgi, whereas PI(4,5)P2 is mainly distributed at the PM and at polarized sites on the cell cortex, in a cell cycle-dependent manner [1,2,3]. As key regulators of polarized exocytosis, the axis of secretory transport from the Golgi to the PM is determined by PI4P and PI(4,5)P2 metabolism within the cell.
Phosphoinositide metabolism lays a path for exocytosis
Saccharomyces cerevisiae serves as a model for understanding the regulation of phosphoinositide metabolism, as it impacts polarized exocytosis. In addition to being a well-established experimental platform for molecular genetics and, historically, a vanguard for understanding mechanisms of membrane trafficking, budding yeast is a model for investigating the dynamic regulation of membrane lipids [4,5,6]. In yeast, many biochemical and genetic techniques can be exploited, including the use of genetically encoded fluorescent lipid probes, some of which were first developed in mammalian systems for in vivo monitoring of lipid and phosphoinositide dynamics [7,8,9,10,11]. Combined, these approaches set the stage for the many extensive investigations of phosphoinositide regulatory activities during yeast polarized exocytosis.
In budding yeast, the spatiotemporal regulation of phosphoinositide metabolism is primarily dependent on the interplay between lipid kinases and phosphatases. The major synthesis of Golgi PI4P in yeast cells is conferred by the type IIIβ PI4-kinase Pik1p, whereas PI4P in the PM is produced by the IIIα PI4-kinase Stt4p [1, 12]. Lsb6p, a related type II PI4-kinase, can partially suppress the inactivation of Stt4p suggesting that Lsb6p contributes to PI4P synthesis at the PM, but Lsb6p activity is mainly associated with endolysosomal trafficking [13, 14]. In fact, unlike other fungal homologues, Lsb6p in Aspergillus nidulans is associated with the Golgi where it replaces the role of Pik1p in apical polarized secretion [15]. In S. cerevisiae, membrane lipids (including PI4P) in the Golgi are packaged into exocytic vesicles and continually transported to the PM, but PI4P pools within the Golgi and PM are distinct and do not intermix [12]. How Pik1p-generated PI4P contributes to PI(4,5)P2 at the PM, even though PI4P apparently ‘vanishes’ from post-Golgi secretory vesicles at the end of their transit represents an unresolved paradox [16, 17]. In yet another seeming contradiction, PI4P generated by Stt4p is largely polarized in the PM to the growing daughter cell even though Stt4p kinase is generally found in patches within mother cells [18,19,20]. Evidently, after PI4P synthesis, other regulatory factors control PI4P levels, cellular distribution, and dynamics within membranes.
In addition to its synthesis, PI4P metabolism is dictated by dephosphorylation as well as conversion of PI4P into polyphosphorylated phosphoinositides, such as PI(4,5)P2. PI4P is dephosphorylated and thereby depleted by the integral membrane protein Sac1p, which is localized in yeast to the ER under normal growth conditions [21, 22]. This localization poses a seeming contradiction given that Sac1p acts on the Stt4p-generated PI4P pool in the PM [21]. However, because Sac1p localizes to membrane contact sites between cortical ER and the PM, Sac1p activity is largely directed at the PM pool of PI4P [19, 23, 24]. Elimination of SAC1 or the tether genes conferring ER-PM membrane contact results in uniform PI4P redistribution throughout the PM where normally PI4P is concentrated at sites of polarized growth at the PM [23, 24]. It should be noted, however, that deleting SAC1 in cells lacking ER-PM contact sites results in lethality, indicating that Sac1p and ER-PM tethers have additional contributions beyond just maintaining PM PI4P levels [25]. In addition to Sac1p, other phosphoinositide phosphatase homologues in budding yeast appear to have wider substrate specificities for phosphoinositides than just PI4P, though they too are significant regulators of cellular PI4P [1, 21, 26]. Nonetheless, Sac1p represents an important determinant of PI4P levels and polarized distribution in the PM, which also affects the distribution of the PI4P derivative PI(4,5)P2 [27].
Stt4p generates PI4P in the PM, which in turn serves as the substrate for PI(4,5)P2 synthesis by Mss4p, the sole PI4P 5-kinase in budding yeast that is also a key determinant of polarized PM growth [18, 27,28,29,30,31,32]. Consistent with its role in controlling phosphoinositide metabolism at the PM, MSS4 (multicopy suppressor of Stt4 mutation) was initially identified as a suppressor of the temperature-sensitive (ts) stt4-1 mutation that affects PI4P pools in the PM [33]. However, neither pik1ts or stt4ts are lethal when combined with an mss4ts allele, suggesting that the Golgi and PM pools of PI4P generated by Pik1p and Stt4p both contribute PI4P for Mss4p activity, as confirmed by the drastic reduction of PI(4,5)P2 in pik1ts stt4.ts double mutant cells [12]. In the PM, PI(4,5)P2 is concentrated at sites of membrane growth but synthesis of PI(4,5)P2 by Mss4p cannot be solely responsible for this polarized distribution; it is debatable whether PI(4,5)P2 has an intrinsic capability to self-form or maintain membrane domains by itself[34, 35]. Rather, the interplay between Mss4p, Sac1p, Stt4p, Pik1p, and lipid transfer proteins dynamically appear to generate localized PI(4,5)P2 membrane concentrations that then recruit PI(4,5)P2-binding effectors. PI(4,5)P2-enriched domains are crucial for the assembly of protein complexes exocytic vesicle targeting as the prerequisite for membrane fusion at sites of polarized growth [36,37,38]
In the dynamic regulation of phosphoinositide-enriched domains within membranes, soluble lipid transfer proteins play an important role in shuttling PI4P between membranes to regulate its turn-over or conversion to PI(4,5)P2 [39, 40]. In particular, the ORP (Oxysterol binding protein [OSBP] related protein) family is implicated in phosphoinositide lipid exchange between different membranes [41]. Yeast ORPs are encoded by the seven OSH (OSBP homologue) genes (OSH1-OSH7), which all share essential functions that any individual OSH performs in the absence of the others [42]. The canonical mammalian OSBP was first shown to change its localization in response to hydroxycholesterol binding, and ORPs were subsequently demonstrated to bind and transfer other lipids, including PI4P and PI(4,5)P2 [43,44,45,46]. ORPs bind sterols and phospholipids, and these lipids are exchanged for phosphoinositides, which then leads to cycles of lipid transfer between membranes. The transfer of phosphoinositides appears to be the common function of all ORPs. Given that collectively the yeast OSHs have shared overlapping essential function/s, it follows that OSHs are important regulators of the essential function of maintaining intracellular phosphoinositide distribution. Indeed, deletion of all OSH genes causes a marked increase in PI4P and PI4P redistribution ubiquitously around the PM [16, 23, 47]. In these cells, the disruptions in phosphoinositide regulation are concurrent with polarization defects affecting cell morphology, septin ring assembly, actin polarization, chitin deposition, and polarized exocytosis [48]. During polarized exocytosis, the archetypal yeast ORP Osh4p (also referred to as Kes1p) tracks along on post-Golgi vesicles as they transit from the Golgi to polarized sites of growth on the PM [49,50,51]. Moreover, Osh4p coprecipitates with regulators and subunits of the exocyst tethering complex [49]. These findings implicate Osh4p, along with other Osh proteins such as Osh6p, in the final events of vesicle targeting to the PM [51]. Indeed, the inactivation of all Osh proteins (but not removal of individual Oshs) causes the accumulation of undocked vesicles in daughter buds and a uniform redistribution of PI4P around the PM [16, 48, 49]. During vesicle biogenesis, however, OSH4 also plays a unique role as nascent vesicles bud from the trans-Golgi on their way to the PM. OSH4 deletion bypasses the essential requirement for SEC14, which encodes a PI/phosphatidylcholine (PC) transfer protein that is key for exocytic vesicle budding from the Golgi [52,53,54,55]. ORP-dependent lipid exchange helps regulate exocytosis by controlling the dynamic distribution of PI4P and PI(4,5)P2 [16, 50, 56], though the role in phosphoinositide regulation does not preclude additional roles for ORPs acting directly on the transport machinery.
PI4P and DAG promote post-Golgi vesicle biogenesis
After protein translocation into the ER and the subsequent delivery of secretory proteins to the late Golgi, the Pik1p-dependent synthesis of PI4P regulates vesicle biogenesis that initiates exocytic trafficking to the PM [12, 21, 53] (Fig. 1A). The production of PI4P by Pik1p is limited by the synthetic availability of its substrates, which is contingent on PI (phosphatidylinositol) transfer from ER to the Golgi [53]. As a PI/phosphatidylcholine (PC) transfer protein, Sec14p is proposed to move PI precursors for PI4P synthesis in the Golgi, which is ultimately essential for post-Golgi vesicle biogenesis [53, 54]. Inactivating Sec14p PI/PC transfer function has the same effect on vesicle biogenesis as eliminating Pik1p [53, 55, 57, 58]. Moreover, Pik1p overexpression in sec14-3 cells can partially rescue growth and secretion defects, but Sec14p overexpression is unable to rescue growth defects of pik1-63 or pik1-83 cells [53]. Thus, these results are consistent with the concept that Sec14 acts upstream to regulate Pik1p generation of Golgi PI4P. Despite that Sec14p transfers PI and PC between liposomal membranes in vitro, mutations that impair Sec14p binding to PI or PC do not disrupt its essential in vivo functions during post-Golgi secretion [58,59,60]. These results suggest that while Sec14p lipid transfer activity may contribute to its overall physiological function, Sec14p impacts vesicle formation as a regulator of phospholipid metabolism and PI4P synthesis in Golgi compartments.
The regulatory role of Sec14p during PI4P-dependent vesicle biogenesis has been revealed through the identification of suppressors that bypass the essential function of SEC14 [47, 61,62,63,64]. The cellular requirement for SEC14 can be entirely abolished by inactivating specific genes encoding phospholipid synthetic enzymes or, through a mechanism affecting Pik1p-generated PI4P, by deleting SAC1 or OSH4/KES1 [61,62,63,64]. These findings suggest that Sec14p links PI4P regulation and phospholipid synthesis. PI-bound Sec14p increases Pik1p-dependent production of PI4P and PI(4,5)P2, and thereby activates Spo14p, a phospholipase D homologue that converts PC to phosphatidic acid (PA) for DAG synthesis [65, 66]. On the other hand, PC-bound Sec14p inhibits choline-phosphate cytidylyltransferase thereby inhibiting PC synthesis and increasing DAG pools [67]. Through both mechanisms, Sec14p activity increases DAG within the Golgi membrane bilayer, which is proposed to affect vesicle biogenesis via membrane deformation and curvature, as well as inducing membrane disorder promoting vesicle scission [68,69,70]. Others, however, question the overall impact of DAG on Golgi membrane curvature [71]. It is curious that Pah1p, the PA phosphatase that produces DAG, has no reported role in the formation of exocytic vesicles from the late Golgi. Regardless, DAG acts in other ways to activate vesicle biogenesis at the trans-Golgi, alongside PI4P [69, 72].
DAG and PI4P metabolism cooperate in the activation of Arf1p (ADP-ribosylation factor 1), a small GTPase regulator of Golgi maturation and post-Golgi vesicle formation [73] (Fig. 1A). Early in the process in vesicle formation, Arf1p is activated by the guanine nucleotide exchange factor (GEF) Sec7p, which acts to recruit more GTP-bound Arf1p to the Golgi membrane thereby creating a positive-feedback loop [74]. Moreover, Sec7p is also an effector of Golgi Rab GTPases including Ypt31p/Ypt32p, which are critical for the regulation and recruitment of later effectors of post-Golgi vesicle transport [75, 76]. Ypt31p/Ypt32p further augments Sec7p GEF activity that, together with increased PI4P levels, drives cargo sorting into vesicles [72, 75]. Sec7p activation of Arf1p also recruits Pik1p to further boost PI4P production; inhibition of either Arf1p or Sec7p results in Pik1p mislocalization and impaired Golgi PI4P production [12, 72, 77]. As shown by in vitro liposome binding, DAG also facilitates GTP-Arf1p recruitment of Pik1p, through in vivo the role of DAG is less clear [77]. In yeast cells, DAG appears to promote vesicle release from the Golgi by stimulating Gcs1p and Age2p ArfGAP activities, which in turn leads to membrane dissociation of GDP-bound Arf1p [78, 79]. In this manner, DAG facilitates Arf1p GTP-to-GDP switching for vesicle biogenesis, enabling Arf1p recycling after membrane scission for subsequent rounds of vesicle formation.
The balancing act controlling Golgi PI4P
As bypass suppressors of SEC14, the deletions of SAC1 and OSH4 appear to impact PI4P incorporation into post-Golgi vesicles by limiting PI4P accumulation within the trans-Golgi [80] (Fig. 1A). Under nutrient-plentiful conditions, yeast Sac1p resides in the ER close to ER-PM contact sites where it primarily affects the Stt4p-synthesized PI4P pool within the PM [25, 81]. Given that Sac1p localization is mainly restricted to the ER, Sac1p lacks a direct means to access Golgi PI4P. Current models involve PI4P transfer from the Golgi to the ER, where PI4P is then hydrolyzed to PI by Sac1p [26, 44, 53]. In mammalian cells, OSBP is recruited to ER-Golgi membrane contact sites (MCSs) where it transfers ER-derived cholesterol to the Golgi and then returns Golgi-synthesized PI4P to the ER, where it is presented to Sac1p [41, 82, 83]. OSBP-dependent increases of cholesterol in the Golgi recruits PI 4-kinases, which represents one mechanism leading to local increases in PI4P [84, 85]. In this manner, OSBP controls the balance between Golgi PI4P synthesis and its turn-over by Sac1 in the ER. Interactions between the OSBP PH (Pleckstrin Homology) domain and Golgi PI4P, and between the OSBP FFAT motif and the ER tether VAP-A, direct OSBP localization to ER-Golgi MCSs [86]. Although the function of yeast Osh4p approximates that of OSBP, Osh4p lacks the sequence elements that confer OSBP recruitment to mammalian ER-Golgi MCSs [39, 41]. However, if ER-Golgi MCSs facilitate Osh4p-dependent PI4P regulation at the trans-Golgi, then elimination of ER-Golgi tethers might also bypass the essential function of SEC14, like SAC1 or OSH4 deletions. Although no known tethers have been reported as SEC14 bypass suppressors, the putative ER-Golgi tether/lipid transfer protein Fmp27 must be present to permit the bypass suppression of sec14∆ when OSH4 or phosphatidylcholine biosynthetic genes are deleted [63]. FMP27 encodes a Vps13p-like tether/lipid transfer protein that localizes to multiple MCSs [87] and might be a candidate tether that links the ER and Golgi together to promote Osh4p lipid transport. Regardless, Osh4p and Sac1p act on Golgi PI4P as a brake to control Sec14p stimulation of Pik1p-dependent PI4P synthesis.

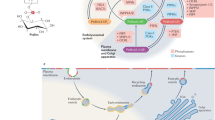
Phosphoinositide regulation of polarized exocytosis from the Golgi to the PM. (A) During Golgi vesicle biogenesis, the balanced exchange of lipids between the ER and Golgi lays the groundwork for PI4P generation. Sec14p reciprocally transfers PC from the trans-Golgi to the ER and PI from the ER to the trans-Golgi. PC-bound Sec14p inhibits DAG production through Pct1p whereas PI-bound Sec14p promotes DAG production via Spo14p. In the trans-Golgi, the Pik1p kinase converts the newly transferred PI to PI4P. In the opposing direction, Osh4p removes Golgi PI4P by transferring PI4P to the ER, where the Sac1p phosphatase dephosphorylates PI4P to PI. Golgi PI4P recruits Rab GTPases Ypt31/32p and indirectly stimulates Sec7p GEF activity, which in turn stimulates Arf1p GTPase activity. Arf1p activates the TRAPPII GEF complex and the Pik1p kinase, which further amplifies PI4P production and Ypt31/32p activity. Golgi accumulation of DAG stimulates Gcs1p and Age2p two Arf GAPs that inactivate Arf1p during later stages of vesicle development. (B) As vesicles are released from Golgi, Ypt31p/32p and PI4P recruit the Rab GEF Sec2p and the type V myosin motor Myo2p for vesicle transport along actomyosin cables towards sites of polarized membrane growth. As a mechanism for Sec2p activation of the Rab GTPase Sec4p, PI4P in the vesicle membrane is removed during vesicle transit. Osh proteins (e.g. Osh4p) facilitate PI4P removal from vesicles either by vesicle-ER lipid exchange, in which Sac1p turns-over PI4P in the ER, or vesicle-PM exchange, wherein PI4P is converted to PI(4,5)P2 by Mss4p in the PM. (C) PI4P removal from vesicles causes Sec2p to swap Ypt31p/32p for Sec4p binding and activation. Sec2p and GTP-Sec4p recruit Sec15p for the assembly of the exocyst complex. On the PM, PI(4,5)P2 clusters the Rho GTPases Cdc42p, Rho1p and Rho3p. During vesicle docking with the PM, the exocyst complex subunits Sec3p and Exo70p bind PI(4,5)P2 and the Rho GTPases. As mediated by Sec6p and Sro7p/77p, tethering by the exocyst complex transitions to membrane fusion as mediated by the Snc1p/2p v-SNAREs and Sso1/2p, Sec9p t-SNAREs
PI4P sorts lipids and proteins during post-Golgi vesicle biogenesis
The accumulation of PI4P in the Golgi leads to local changes in lipids, secretory cargo sorting, and membrane coat formation that elicit exocytic vesicle biogenesis. In addition, membrane distortions generate bilayer curvature to initiate vesicle budding from the Golgi. Drs2p is a type IV P-type ATPase “lipid flippase” that translocates phosphatidylserine (PS) and phosphatidylethanolamine (PE) across the membrane bilayer to the cytoplasmic leaflet and establishes membrane asymmetry in the trans-Golgi [88, 89]. Drs2p flippase activity is activated by PI4P [90, 91], and Drs2p-generated membrane subdomains primes membrane curvature for shaping into exocytic vesicles by coat proteins [89, 92,93,94,95]. Through Drs2p flippase activity, clathrin facilitates the formation of post-Golgi vesicles and aides exocytic cargo sorting [89, 92, 94]. Drs2p-dependent PS leaflet flipping is activated by Pik1p, and Osh4p action opposes Pik1p, therefore Drs2p and Osh4p have mutually antagonistic activities [89,90,91, 96]. In addition, Osh4p-dependent depletion of Golgi PI4P reduces Drs2p activity [89,90,91, 96]. The dynamic equilibrium of Golgi lipid composition as controlled by Drs2p and Osh4p generates the membrane architecture imparted by the lipids found in exocytic vesicles budding from the Golgi [97].
As a downstream consequence of Pik1p and Sec14p activities, with membrane conditions set by lipid recruitment and PI4P signaling, protein sorting and cargo accumulation is dictated by Arf1p regulation of clathrin and exomer adapter complexes [54, 66, 98, 99] (Fig. 1A). The direct (endosome-independent) route to the PM is defined by lower-density post-Golgi vesicles containing the secreted endoglucanase Bgl2p, which is a marker for polarized exocytosis to sites of membrane growth in daughter buds [100]. Bgl2p polarized exocytosis is dependent on Sec14p and Osh proteins where the polarized targeting of vesicular traffic to regions of active growth ensures the accumulation of protein cargo necessary for surface expansion [48, 54, 100]. In addition to the sorting of Bgl2p with other cargo destined for polarized sites, nascent vesicles are loaded with the transport machinery necessary for post-Golgi vesicle transit, docking and fusion with the PM [101]. The coordination of the next post-Golgi steps in exocytic vesicle trafficking are also regulated by phosphoinositides, which affect the sequential activation of several different small GTPases whose activities control specific transport events [17, 102, 103]. The Pik1p-dependent generation of PI4P induced by Sec7p, Arf1p, and DAG ensures recruitment of factors into post-Golgi vesicles needed later during vesicle transport to the PM. Sec7p activation of Arf1p recruits the TRAPPII GEF complex to the trans-Golgi to induce the Ypt31/32p Rab GTPases [2, 75, 104]. GTP-Ypt31/32p further activates Sec7p, which along with Pik1p-generated PI4P, leads to the assembly of essential factors for the final steps of vesicle budding from the trans-Golgi [2, 105]. At sites of vesicle budding, GTP-Ypt31/32p and PI4P recruit the Rab GEF Sec2p, a crucial regulator of later stages in exocytosis [106,107,108]. Sec2p is not a GEF for Ypt32p, rather the Sec2p-Ypt32p interaction only serves to attach Sec2p, along with its binding to PI4P, to nascent post-Golgi vesicles [107, 108]. The Sec2p GEF is, however, required for Sec4p Rab GTPase activation that designates the next event in the regulatory cascade of small GTPases during polarized exocytosis [107] (Fig. 1B). On nascent vesicles, Sec4p, Ypt31/32p, and PI4P also bind the type V myosin Myo2p to initiate the general movement of exocytic vesicles along actin cables to sites of polarized growth on the PM, as detailed below [109,110,111].
PI4P facilitates vesicle maturation during transit and along actin/myosin cables
In addition to directing post-Golgi vesicle transit to the PM, Myo2p affects the final mechanical steps in vesicle release from the trans-Golgi (Fig. 1B). Myo2p can induce membrane curvature and vesicle budding by pulling on the membrane [112], though some vesicles appear to be released from the Golgi without Myo2p action [113]. The initial Myo2p-dependent attachment of exocytic vesicles, and their continued association with actin cables, appears to be at least partly dependent on PI4P incorporation into vesicle membranes. Sec4p and Ypt31/32p GTPases act together to bind vesicles to myosin-associated actin transport cables, but the necessity for Myo2p-Rab GTPase interactions can be bypassed by enhancing Myo2p interaction with PI4P [110]. Consistent with these results, increasing Golgi PI4P using sac1Δ or PIK1 overexpression rescues the defective localization of myo2-12p at its restrictive temperature, underscoring the importance of PI4P in Myo2p localization [110]. In the timeline of exocytic vesicle transit, Ypt31/32p remains on vesicles after budding from the Golgi whereas both Sec2p and Sec4p co-localizes with Ypt31/32p immediately prior to vesicle release [113]. In close succession, Myo2p localizes on budding vesicles after Sec4p, suggesting that Myo2p is immediately recruited upon Sec4p activation [113]. In addition to the Rab GTPases and PI4P interactions with Myo2p, Myo2p also interacts with Sec15p, a subunit of the exocyst tethering complex that establishes first contact between exocytic vesicles and the PM [111, 114]. The current model envisages Ypt31/32p first capturing Myo2p as vesicles bud from the trans-Golgi after which Ypt31/32p is swapped with GTP-Sec4p and Sec15p, which both bind Myo2p until transport nears completion [111]. Through the interaction with Sec15p, Myo2p indirectly binds and affects other exocyst complex components, perhaps facilitating the assembly of the exocyst tether complex.
PI4P regulation of Sec2p is the key spatiotemporal regulator of exocyst complex assembly
One of the most consequential effects of PI4P on post-Golgi vesicle trafficking to the PM involves the recruitment and regulation of the Sec2p GEF. During vesicle budding from the Golgi, Sec2p binds Ypt31p/32p and PI4P on nascent post-Golgi vesicles [106,107,108] (Fig. 2, Middle). Sec2p has three low-affinity PI4P binding sites that appear to play a direct role in regulating Sec2p activation of small GTPases and effector interactions [108]. These PI4P binding sites map to three positively charged patches inside predicted loops region towards the C-terminal end of Sec2p, and the PI4P-binding patches are collectively required for Sec2p localization to vesicles [108] (Fig. 2, Middle). Sec2p is mislocalized in pik1-101 cells at their restrictive temperature, confirming the dependence of Sec2p recruitment on Golgi PI4P production [108]. Due to the relatively low specificity of these charged patches, Sec2p can be recruited by other negatively charged lipids, albeit to a lesser extent [108]. Although PI4P and Ypt31/32p recruit Sec2p to vesicles, PI4P does not affect the binding of Ypt31/32p to Sec2p [108]. Thus, Sec2p recruitment to vesicles involves co-incidence detection of both Ypt31/32p and PI4P.

AlphaFold3-predicted Sec2p domains [242] involved in regulator and effector binding. (Top) Predicted structure of homodimeric full-length Sec2p shown with colouring corresponding to predicted local distance difference test (pLDDT) per-residue confidence scores; disordered C-terminal regions shown as simple lines. Residue-residue alignment confidence is shown for the Sec2p homodimer in the predicted aligned error (PAE) plot. (Middle) Sec2p domain interactions during vesicle biogenesis. Sec2p(160–258) region (green) binding Ypt31/32p and Sec2p(421–456) basic patches (magenta) binding PI4P. Binding domains shown as space-filling representations and other regions shown as ribbon diagrams. (Bottom) Sec2p domain interactions during Sec4p activation and initiation of exocyst complex assembly. Sec2p(60–160) region (yellow) binding Sec4p, Sec2p(160–258) region (light blue) binding Sec15p, and Sec2p(S181, S186, S188) phosphorylated serine residues (red). Binding domains shown as space-filling representations and other regions shown as ribbon diagrams. The Sec2p PI4P-binding and phosphorylation sites are positioned between the Sec2p GEF (Sec4p interaction) helical domain and the “regulatory” (Ypt31p/32p and Sec15p interaction) globular domain
Ypt31/32p and PI4P recruit Sec2p to nascent vesicles, but these interactions inhibit the GEF function of Sec2p during the activation of the Sec4p small GTPase, which is a key regulator of later exocytic events [108] (Fig.1B). Sec2p does not act as a GEF for the Ypt31/32p small GTPases, despite their physical interaction [107]. Rather, Ypt31/32p binding to Sec2p adjacent to the Sec2p GEF domain blocks Sec15p binding [108, 115] (Fig. 2, Bottom). 1 The transition of Sec2p binding of Ypt31/32p to the GEF activation of Sec4p and Sec2p-Sec15p binding is attendant with the removal of PI4P from the transiting vesicles and from Sec2p itself [102, 108].
Ypt31/32p and PI4P not only affects Sec2p GEF activation of Sec4p, but they also inhibit Sec2p interactions with Sec15p, a component of the exocyst complex recruited by GTP-Sec4p [26, 108, 116]. Ypt31/32p competes with Sec15p for binding to the same domain of Sec2p [108, 117] (Fig. 2). Upon depletion of PI4P from post-Golgi vesicles, the Sec2p GEF activates Sec4p binding and favours Sec15p binding over Ypt31/32p. After PI4P removal, the complex of GTP-Sec4p, Sec15p, and Sec2p then elicits the initial events of exocyst complex assembly [108, 113] (Fig. 1C). In the octameric exocyst complex, Sec15p forms a tetrameric subcomplex helical bundle with Sec10p, Exo70p and Exo84p [116]. The Sec15p nucleated subcomplex then makes extensive interfaces with the other exocyst tetrameric subcomplex, formed by Sec3p-Sec5p-Sec6p-Sec8p, which generates the holo-exocyst complex. Given its length, Sec15p has a particularly important role as a scaffold to unite subcomplexes on vesicles, which affects the assembly of the entire exocyst complex [116]. In concert with the small GTPases Rho1p, Rho3p and Cdc42p, and the lethal giant-larvae homologues Sro7/77p at the PM, the exocyst complex then tethers vesicles to the PM and permits ensuing SNARE-mediated membrane fusion [108, 113, 118] (Fig. 1C). Thus, as the trigger for Sec2p regulatory activity, reduction of PI4P levels from post-Golgi vesicles is key to initializing exocyst complex docking upon vesicle arrival at the PM.
To explain how PI4P might be depleted from vesicle membranes while en route to the PM, it was proposed that the PI4P is removed by Osh proteins in their capacity as soluble phosphoinositide lipid transfer proteins [39, 108] (Fig. 1B). The removal of PI4P from vesicles, and PI4P dissociation from Sec2p, promotes Sec2p phosphorylation by the casein kinases Yck1p/2p thereby triggering Sec2p-Sec4p-Sec15p binding, and exocyst complex assembly [108, 117]. The Yck1p/2p phosphorylation sites lie within the Sec2p domain defined by the mutually exclusive binding of Ypt32p and Sec15p [117] (Fig. 1). The Yck2p kinase binds the Sec2p autoinhibitory domain adjacent to its PI4P binding pockets; PI4P binding inhibits Sec2p phosphorylation [117]. The proposed role of Osh4p in removing PI4P from post-Golgi vesicles, and thereby from Sec2p, is consistent with the finding that OSH4 deletion inhibits Sec2p-Sec15p binding [39, 50, 117]. Furthermore, the combined deletions of both YCK1 and OSH4 cause severe growth defects in keeping with functional interaction between Osh4p and Sec2p phosphorylation [119]. Thus, PI4P extraction from the exocytic vesicles appears to be an essential event for the final steps of exocytosis at the PM.
Osh proteins deplete PI4P from the vesicle membrane to promote exocyst complex assembly
In addition to unique affinities for either sterols or PS, all Osh proteins appear to bind phosphoinositides, and collectively they are required for phosphoinositide metabolism and regulation of polarized secretion [16, 20, 23, 39, 49, 51, 120]. Nonetheless, Osh4p remains the focus of study for determining Osh protein activities during vesicle-PM tethering, which is distinct from the unique role of Osh4p during Sec14p-dependent Golgi vesicle biogenesis [48, 49] (Fig. 1). After vesicle biogenesis, Osh4p remains on post-Golgi vesicles during their entire transit to the PM [49,50,51]. Osh4p co-precipitates with multiple subunits of the completely assembled exocyst complex, including the PM- and vesicle- associated small GTPases Cdc42p, Rho1p, and Sec4p [49]. Moreover, in cells where Osh proteins have been inactivated, Cdc42p and Rho1p polarization on the PM is disrupted and Sec4p accumulates on undocked exocytic vesicles [48]. Multicopy OSH4 suppresses CDC42 mutations that specifically affect polarized exocytosis and OSH4 overexpression re-establishes the polarized localization of the corresponding mutant Cdc42 proteins [48]. These findings suggested that through their lipid exchange activities, Osh4p and other Osh proteins remove PI4P from vesicles and thereby promote exocyst complex tethering during vesicle docking with the PM [39].
Given that Osh4p transfers PI4P and binds to the entire assembled exocyst complex, two possibilities are suggested: (1) Osh4p is recruited to nascent secretory vesicles by PI4P and exocyst complex subunits where, as a soluble lipid transfer protein, Osh4p removes and transports PI4P from post-Golgi vesicles to the ER for PI4P dephosphorylation by Sac1p [39, 50]; (2) Osh4p is recruited to the vesicle by PI4P and exocyst complex subunits where Osh4p transfers PI4P from vesicles to the PM for PI4P conversion to PI(4,5)P2 by the PI4P-5-kinase Mss4p (Fig. 1B). In either case, Osh4p physical association with components on the assembled exocyst complex suggests that vesicular PI4P depletion by Osh4p does not initiate but rather maintains exocyst complex assembly. Even though Osh4p is continuously associated with transiting post-Golgi vesicles, PI4P dissipates from vesicles only when in the vicinity of the PM [16, 49,50,51]. Consistent with findings that myosin association with vesicles is partially dependent on vesicle PI4P, PI4P removal at the end of exocytosis is concomitant with Myo2p detachment from vesicles upon arrival at the PM. In addition to how Osh proteins affect vesicular PI4P, Osh proteins also impact PI(4,5)P2 levels on the PM given that PI(4,5)P2 levels are reduced in cells following Osh protein inactivation [23, 56]. It is not clear whether Osh proteins somehow make the Pik1p Golgi/vesicle pool of PI4P available for PI(4,5)P2 production by Mss4p. During vesicle docking at the PM, the close proximity of two membranes might facilitate Osh4p sterol/PI4P exchange thereby providing PI4P to fuel Mss4p production of PI(4,5)P2 production in the PM. Nonetheless, because Osh4p removes PI4P from vesicles it is a key driver in maintaining exocyst complex stability, which involves continued activation of Sec4p by its GEF Sec2p.
PI(4,5)P2 at sites of polarized growth in the PM recruits regulators of exocytic vesicle docking and membrane fusion
In both yeast and metazoan cells, the final target site for polarized exocytosis on the PM is delimited by PI(4,5)P2-enriched areas where exocytic vesicles dock prior to membrane fusion and cargo delivery [38, 121, 122]. On the PM, the generation of PI(4,5)P2 by Mss4p affects the targeting of post-Golgi vesicles by affecting the polarized positioning of actin cables, which determines the direction of vesicle transit, and by amassing small GTPases on the PM to enable exocyst complex tethering [29] (Fig. 1C). Secretory vesicles are transported along actin cables near sites of polarized cell growth at the PM; disruption of actomyosin cables inhibits polarized exocytosis and causes PM defects that lead to isotropic cellular growth [3, 12, 21, 29, 123,124,125]. The Rho-like small GTPase Cdc42p interacts with upstream and downstream effectors to nucleate the actin cytoskeleton and, in turn, polarized exocytosis delivers Cdc42p to the PM to maintain membrane polarization [28, 126, 127]. The main Cdc42p GEF, Cdc24p, establishes cell polarization that ultimately promotes polarized exocytosis. However, due to its low affinity phosphoinositide-binding PH domain, the role of Cdc24p in polarized exocytosis is largely PI(4,5)P2-independent [128]. Other PI(4,5)P2-binding effectors such as Gic1p/2p, however, bind PI(4,5)P2 and activate Cdc42p during polarized exocytosis and polarized growth [126, 129,130,131]. In addition to Cdc42p, Rho3p plays dual roles in both the polarized positioning of the actomyosin cytoskeleton and in exocyst complex tethering; Rho3p directly interacts with Myo2p and the exocyst complex component Exo70p[132, 133]. Exo70p binds both Rho3p and PI(4,5)P2, like Cdc42p effectors that bind PI(4,5)P2 [134]. The third Rho GTPase involved in exocyst complex tethering to the PM, Rho1p, is also directed to polarized membrane sites by PI(4,5)P2 [135]. While Cdc42p is important in establishing cellular polarization, Rho1p and Rho3p appear to be necessary for its maintenance. Although Rho1p signaling controls actin organization, direct physical interaction between Rho1p and the actomyosin cytoskeleton has not been reported [136]. Nonetheless, PI(4,5)P2 acts alongside Cdc42p, Rho1p, and Rho3p to interact with exocyst complex components to receive vesicles at sites of polarization on the PM.
Rho GTPases ensure proper targeting of post-Golgi vesicles through direct interactions with specific “landmark” exocyst complex components, though vesicle docking is further augmented by PI(4,5)P2 interaction [30, 118, 137,138,139,140,141,142] (Fig.1C). As landmark exocyst complex subunits, Sec3p directly binds Rho1p whereas Exo70p interacts with both Cdc42p and Rho3p [30, 143]. The Sec3p PH domain and polybasic surface patches at the C-terminus of Exo70p also enhance association with the PM via PI(4,5)P2 binding [30, 38, 144]. Indeed, increased PM PI(4,5)P2 production by MSS4 kinase overexpression can rescue growth defects of SEC3 and EXO70 mutants [3, 30, 144]. The metazoan Exo84p homologue interacts with phosphoinositides through an atypical PH domain. However, yeast Exo84p does not contain a PH domain and PI(4,5)P2 interactions with the yeast exocyst complex at the PM appears to be restricted to Sec3p and Exo70p [145, 146].
Unlike most yeast exocyst complex subunits, interactions between the mammalian subunits are more dynamic given that components neither arrive concurrently nor remain together at exocytic fusion sites [147, 148]. However, even in yeast there is some question of when and where some components assemble with the entire exocyst complex. Yeast Sec3p and Exo70p can be targeted to the PM independently of actin/Myo2p-mediated vesicle transport, but Sec3p and Exo70p appear to be stably associated with the entire octameric exocyst complex during vesicle transit to the PM [144, 149, 150]. Adding to this perplexity, the absolute necessity of Sec3p is questionable, even though it represents a landmark exocyst complex subunit. At lower temperatures yeast cells can survive if SEC3 is deleted, suggesting that Sec3p affects the efficiency of vesicle docking whereas Exo70p is the essential effector for exocyst complex tethering [31, 118, 131, 151]. Regardless, the interactions between Sec3p, Exo70p, and the clustering of Rho GTPases and PI(4,5)P2 at the PM, initiate the final structural changes necessary for vesicle/PM tethering [132, 138, 142].
Successful vesicle tethering to the PM leads to SNARE-mediated membrane fusion, which is cooperatively facilitated by specific exocyst complex subunits, SNAREs, alongside other regulatory factors. In yeast, the tethering to fusion transition is partially mediated by interactions between Sec6p and Sec3p with the vesicle-bound SNAREs Snc1/2p and the PM target-SNAREs, Sec9p and Sso1/2p [116, 152,153,154,155] (Fig. 1C). Furthermore, the interactions of Sro7p/Sro77p with Sec4p, Exo84p and the t-SNARE Sec9p act in parallel to successfully integrate vesicle tethering and fusion with the PM [118, 139, 140, 156,157,158]. Functional interactions between SEC9 and MSS4 suggest that PI(4,5)P2 impacts SNARE-mediated membrane fusion during polarized exocytosis, though it is unclear how [3, 121, 159]. It should be noted that during the specialized vesicular fusion generating prospore membranes during yeast sporulation, PI(4,5)P2 binding appears to drive Sso1p (but not Sso2p) conformation changes that specifically activates Sso1p assembly with other SNAREs [160]. However, as a general mechanism, the role of phosphoinositides in membrane fusion during yeast polarized exocytosis remains an open area for future study.
Budding yeast as a simple model for understanding the impact of phosphoinositide regulation during polarized exocytosis in human diseases
Molecular mechanisms determined from analyzing phosphoinositides in S. cerevisiae have direct implications for the therapeutic targeting of polarized exocytosis during host invasion by pathogenic fungi. Non-pathogenic filamentous fungi such as Neurospora species and opportunistic pathogenic yeast such as Candida albicans can grow by polarized extensions of hyphae, which represent expansions of the PM supplied by a constant transport of secretory vesicles from the Golgi [161]. In C. albicans, Golgi Pik1p-generated PI4P is necessary for the transition to filamentous growth due to its requirement for vesicle biogenesis and targeting to sites of growth at the PM [162]. Unlike S. cerevisiae, however, in C. albicans the PM synthesis of PI4P by the Stt4p kinase is not essential for cell viability, but the PM PI4P pool is still important for hyphal morphogenesis (just not vesicle trafficking per se) [163, 164]. Although not specifically tested in the context of polarized exocytosis, C. albicans Mss4p is localized at filament tips where it increases the localized concentration of PI(4,5)P2, which is necessary for hyphal formation and virulence [165, 166]. However, because the PM pool of PI(4,5)P2 in C. albicans appears normal in the absence of PM Stt4p-generated PI4P, it has been proposed that Golgi PI4P still reaches the PM even though Golgi and PM PI4P pools are separate and distinct [162,163,164]. Similar to what is hypothesized in S. cerevisiae, perhaps C. albicans Osh proteins also transfer PI4P from vesicles to the PM for rapid PI(4,5)P2 conversion by Mss4p at the PM. If so, C. albicans Osh proteins might be efficacious targets for blocking hyphal growth through the inhibition of PI4P transfer to the PM. As small molecule inhibitors of ORPs, and potentially of C. albicans Osh proteins, ORPphilins might be effective in disrupting PM phosphoinositide metabolism to prevent hyphal infection [167].
The phosphoinositide-dependence of hyphal growth is pertinent to both pathogenic as well as non-pathogenic fungi, suggesting that PI4P and PI(4,5)P2 might be ubiquitously important for polarized exocytosis during filamentation. For instance, the MSS4 homologue in Neurospora crassa is also necessary for hyphal filamentation, and PI(4,5)P2 synthesis at the PM is important for polarized membrane growth [168]. The general requirement of PM PI(4,5)P2 during fungal hyphal morphogenesis appears to involve coordinating exocytic transport to control the balance between rapid hyphal elongation and the maintenance of polarity at hyphal tips [169, 170]. Although well conserved from fungi to humans, PI transfer proteins with homology to S. cerevisiae Sec14p offer another target for inhibiting fungal Golgi-synthesized PI4P to block virulence factor secretion and the transition to filamentous growth [171,172,173]. It has been proposed that structural subtleties in Sec14p homologues can be exploited to develop small molecular inhibitors with binding selectivities permitting effective antifungal activity without off-target toxicity [174, 175].
As mechanistically defined in S. cerevisiae, the conserved exocyst complex and its regulation via interactions with phosphoinositides might also contribute to polarized exocytosis necessary for hyphal formation in filamentous fungi (Table 1) [165, 176,177,178,179,180]. Unlike S. cerevisiae, however, polarized exocytosis to hyphal growths on the PM proceeds through a subapical intermediate of clustered Sec4p-associated vesicles called the Spitzenkörper [181, 182]. Although the Spitzenkörper does not confer directionality to vesicle trafficking per se, subsequent exocyst complex targeting of secretory transport supports the polarized growth of hyphae at the PM [176, 177, 183, 184]. In terms of potential phosphoinositide-binding proteins, the Sec3p homologue in Candida is required for hyphal formation, as is the Sec2p homologue [185, 186]. At the PM, Rho GTPases is generally required for polarized membrane growth of hyphal filaments, which is partly affected by phosphoinositides [187,188,189,190,191]. In Candida, however, the polarized localization of Cdc42p is independent of PI(4,5)P2, but active Rho1p recruitment to growing hyphal filament is dependent on Stt4p and Mss4p synthesis of PI4P and PI(4,5)P2 at the PM [190]. If mechanistically akin to exocyst complex tethering in S. cerevisiae, phosphoinositide regulation of PM Rho family GTPases is likely an important determinant of polarized exocytosis during hyphal filamentation.
Apart from hyphal formation and fungal pathogenesis, the regulation of phosphoinositide metabolism during polarized exocytosis can impact human disease, which in some cases can be intuited through the conserved mechanisms as originally defined in S. cerevisiae. Defects in phosphoinositide metabolism and regulation are associated with specific cancers, neuromuscular disease and neurodegenerative disorders, including Parkinson’s disease and amyotrophic lateral sclerosis (ALS) [192, 193] (Table 1). While phosphoinositide dysregulation has been directly linked to disorders affecting endolysosomal trafficking or synaptic vesicle exocytosis, few diseases associated with defective polarized exocytosis can be directly traced to changes in phosphoinositide metabolism. For example, metastatic loss of cell–cell adhesion and cell migration is promoted by increased Golgi PI4P, which is otherwise attenuated by the Sac1 inositol 4-phosphatase [22, 194]. However, Sac1 itself has yet to be directly linked to a specific human disease, let alone to one caused by polarized exocytosis defects [195]. In contrast, the dual inositol 4- and 5-phosphatases represented by the Sac1 domain-containing synaptojanins affect other membrane trafficking pathways directly impacting disease [192, 193, 195]. Mutations in the Synaptojanin-1 Sac1-domain disrupts endocytosis and cilia regulation and causes early-onset Parkinson’s disease [196, 197]. Although it lacks a Sac1-domain, OCRL is another inositol 5-phosphatase that hydrolyzes PI(4,5)P2 in the ciliary axoneme, and is linked to a ciliopathy causing Lowe’s syndrome [198] (Table 1). OCRL teams up with Sac2/INPP5F, a Sac1-domain containing inositol 4-phosphatase, to dephosphorylate PI(4,5)P2 akin to the dual phosphatase activities of synaptojanins (and in mice Sac2/INPP5F knock-out synergistically aggravates Synaptojannin-1-related Parkinson’s disease defects) [196, 199]. Because endocytosis and cilia regulation require PI(4,5)P2 dephosphorylation at the PM, whereas polarized exocytosis requires local accumulations of PI(4,5)P2, synaptojanin inactivation might even bolster PI(4,5)P2-dependent polarized exocytosis. Indeed, the deletion of yeast Sac1-domain synaptojanins encoded by INP51-INP53 perturbs endocytosis, but not exocytosis; though synaptojanin overexpression might perturb polarized exocytosis by reducing PM PI(4,5)P2 [1]. In the specific cases of synaptic vesicle trafficking and regulated exocytosis, the generation of both PI4P and PI(4,5)P2 have important functions that impact the pathology of neurodegenerative disease and mental disorders [200,201,202,203]. However, in diseases affecting polarized membrane trafficking, a direct mechanistic link for many PI4P and PI(4,5)P2 kinases and phosphatases is less established. In yeast, overexpression of the Mss4p PI4P 5-kinase rescues defects in polarized exocytosis caused by mutations in CDC42, MYO2, and genes encoding exocyst subunits [3, 121]. Human homologues of Mss4p (PIP5K1C and PIP4K2A/PIP5K2A) are implicated in fetal neural/arthrogryposis defects and schizophrenia, respectively [204,205,206] (Table 1). In terms of PI4P generation, defects in human PI 4-kinase (PI4K) IIIα are associated with the neurological disease hypomyelinating leukodystrophy [207, 208]. In flies, PI4K IIIα defects disrupts the recruitment of the exocyst complex subunit Sec5 [37]. Although it is unclear whether polarized exocytosis is specifically compromised in human phosphoinositide kinase-associated diseases, membrane trafficking to the PM might be an aggravating factor. Analogous to yeast, activation of phosphoinositide kinases might also rescue disease-causing defects in human exocyst complex subunits and regulators. Of the many pharmaceutical targets of PI4P and PI(4,5)P2 signaling, key effector complexes of polarized exocytosis might represent opportunities for therapeutic manipulation [209].
The mammalian exocyst complex drives selective secretion to polarized sites on the PM [210,211,212], but the exocyst complex also mediates constitutive exocytosis in mammals [213]. As such, the exocyst complex is required for neurite outgrowth during neuronal development and membrane dynamics, despite that the exocyst complex is not involved in synaptic transport and neurotransmitter release [214,215,216]. The exocyst complex is important for targeting secretory cargo to cilia, which affects the cellular detection of sensation and extracellular signaling [217, 218]. Mutations in many exocyst complex components also inhibit epithelial polarization in mammalian cells by disrupting transport desmosomes and adherens junctions [219, 220]. As a result, defects in specific components in the exocyst complex are causative agents of neurodegeneration and ciliopathies [211, 212] (Table 1). Defects in the human homologues of yeast SEC5 (corresponding to human EXOC2), SEC6 (EXOC3L2), SEC8 (EXOC4), SEC15 (EXOC6B), EXO70 (EXOC7), EXO84 (EXOC8), and RHO3 (RALA) are associated with a variety of neurodegenerative and neurodevelopmental disorders [212]. Given the mechanistic conservation between yeast and humans, it is not surprising that many mutations that disrupt yeast polarized exocytosis cause related defects in humans manifesting in disease.
As with the yeast exocyst complex, the corresponding metazoan components and effectors share a similar dependence on phosphoinositide regulation. Like in yeast, the interaction between PI(4,5)P2 and the mammalian Exo70 homologue (ExoC7) confers PM association [30, 221, 222]. In addition to ExoC7, several mammalian and yeast exocyst subunits contain phosphoinositide-binding pleckstrin homology (PH) domains or other PI4P/PI(4,5)P2 interacting motifs (Table 1). Mutations in these domains and motifs can disrupt binding to phosphoinositides and can have disease-related consequences. A patient afflicted with Joubert syndrome ciliopathy is homozygous for a mutation within the phosphoinositide-binding PH domain of ExoC8, underscoring the disease relevance of exocyst complex-phosphoinositide interactions [223]. This is consistent with the consequence of losing all EXOC6B function, which results in ciliopathy that causes nervous system and joint impairment [224]. Although not directly linked to any named diseases, mammalian EXOC5 (corresponding to yeast SEC10) and Cdc42 cooperatively interact in the same pathway during ciliogenesis [225,226,227]. In yeast, SEC10 mutations are rescued by overexpression of the PIP4 5-kinase encoded by MSS4, and Cdc42p localization to the PM is dependent on Mss4p function [3, 121]. Thus, based on the yeast findings, the generation of PI(4,5)P2 might also affect the regulation of the ciliogenesis pathway by EXOC5 and Cdc42. Another significant factor in ciliogenesis regulation involves the human homologue of SEC2, encoded by Rabin8 [228]. Rabin8 activation of the Rab8 GTPase (the human homologue of yeast Sec4p) is blocked by PTEN-induced kinase 1 (PINK1), an autosomal recessive agent of Parkinson’s disease [229], suggesting a link between Parkinson’s disease and Rab8 activation by Rabin8. If the yeast Sec4p-Sec2p regulatory mechanism applies to the Rab8-Rabin8 interaction, then vesicular PI4P might impact the progression of Parkinson’s disease. The complex interactions between exocyst complex subunits, their regulators, and both PI4P and PI(4,5)P2 provides therapeutic opportunities for neurodegeneration and ciliopathies. In addition, cancer is also affected by polarized PM trafficking that might be treated via phosphoinositides.
Because the exocyst complex impacts polarized membrane growth, and cancer is often driven by changes in cell polarization, recent findings are consistent with the predication that the exocyst complex directly impacts tumorigenesis [230] (Table 1). For example, knockdown of EXOC2 or EXOC8 inhibits Ras-dependent tumorigenic human cell growth. ExoC2 and ExoC8 act as direct effectors of RalA and RalB GTPases, which in turn are Ras effectors liked to several cancers [231,232,233]. Although Ral GTPases are not present in yeast [146, 234], CDC42 and RHO3 Rho family GTPases functionally interact with yeast SEC5 and EXO84, suggesting a loose mechanistic conservation [118, 119, 139, 235]. In humans, RalA interactions are thought to represent an alternate pathway for exocyst complex activation causing vesicle clustering near the PM, which is predicted to boost subsequent membrane fusion events [118, 146]. In this regard, the RalA GTPase interaction with ExoC8 (i.e. human EXO84) has been proposed to functionally emulate the interaction of Sro7p with Exo84p in yeast [118, 145]. Furthermore, phosphoinositides appear to regulate the Ral GTPase-ExoC8 interaction [146]. The Ral GTPase binding site overlaps with the ExoC8 PH domain, and RalA competes with phosphoinositides for ExoC8 binding. The human homologue of yeast Sro7p, human lethal (2) giant larvae (Hugl), affects polarized exocytosis and is directly implicated in tumorigenesis [156, 236], suggesting that the yeast Sro7p-Exo84p interaction might also have implications for cancer. Because of the physical and functional interconnectivity of all exocyst complex subunits, targeted changes in PI4P or PI(4,5)P2 binding or phosphoinositide metabolism might offer new treatment options for tackling tumorigenesis [209].
Conclusions
The effects of phosphoinositide signaling during polarized exocytosis are focused on specific regulators that mediate transitions between mechanistic steps. Phosphoinositides help drive these transitions by promoting the switching of one small GTPase for the activity of another in the regulatory cascade that defines the overall process of exocytosis [17]. Vesicle biogenesis is dependent on PI4P induction of the Arf1p GTPase and then, in preparation for membrane tethering to the PM, exocyst assembly requires PI4P removal from post-Golgi vesicles for Sec2p GEF activation of the Sec4p GTPase. In parallel, the targeting of vesicles to sites of membrane growth on the PM is dictated by Cdc42p and Rho GTPase polarization by PI(4,5)P2. PI(4,5)P2 further supports exocyst complex interactions with the PM through direct interactions principally with Exo70p. The control of each phosphoinositide regulatory node is conferred by the dynamic actions of lipid transfer proteins (i.e. Sec14p and Osh4p) acting in concert with agents of PI4P and PI(4,5)P2 metabolism (i.e. Sac1p, Stt4p, Pik1p, and Mss4p).
During Golgi vesicle biogenesis, Arf1p activation is intimately coupled to PI4P metabolism and vice versa. PI4P synthesis by Pik1p leads to Ypt31p/32p GTPase recruitment that induces the Arf1p GEF, Sec7p (Fig. 2). PI4P synthesis by Pik1p is further increased by Arf1p activation, which ultimately drives a positive feedback loop for ensuring Arf1p completion of vesicle release [77]. The mechanism controlling this Arf1p feedback loop is less understood. PI4P synthesis is kept in check by Osh4p and Sac1p turn-over of PI4P, and deletion of OSH4 and SAC1 bypasses the essential requirement for Sec14p, just like eliminating the Arf1 GAPs, Age1p and Gcs1p [79]. Gcs1p also binds PI4P in vitro (though Gcs1p has a general affinity for monophosphorylated phosphoinositides) [237], which suggests that Gcs1p might be recruited to the Golgi when high PI4P levels to break the Arf1p positive feedback loop. In addition, Age2p contains a C-terminal domain amphipathic helix that mediates Age2p binding to membranes in vitro [238]. It is a reasonable prediction that PI4P promotes Age2p membrane interactions via this region, in a manner akin to Gcs1p-PI4P binding.
Given its central importance in driving the mechanistic transition from vesicle biogenesis to vesicle targeting, Sec2p demands special consideration. During the transit of post-Golgi vesicles to the PM, PI4P acts directly on Sec2p to dictate when the Sec4p GTPase is activated and when Sec15p binding initiates exocyst complex assembly. The dynamics of PI4P exchange on post-Golgi vesicles is therefore key to changing Sec2p activity states. The depletion of PI4P from vesicle membranes by Osh4p and other yeast Osh proteins represents an important trigger for vesicle docking. It remains an open question as to what controls the timing of PI4P removal by Osh proteins and, ultimately, where this PI4P is transferred.
Whereas PI4P is the primary phosphoinositide impacting vesicle regulation, PI(4,5)P2 in the PM determines where vesicles will dock. The contribution of PI(4,5)P2 to Cdc42p polarization and, by extension, to polarized exocytosis has been enigmatic. Initial Cdc42p polarization is conferred in part by its GEF Cdc24p, which in turn seems to be polarized through cooperative binding to anionic lipids via a PH domain that has very low affinity for phosphoinositides [128]. Once the polarity axis is established, the Cdc24p-Cdc42p interaction is PI(4,5)P2-independent, but other interactions that maintain Cdc42p polarization are affected by PI(4,5)P2 [3]. Several Cdc42p and Rho GTPase effectors/regulators have PH domains or polybasic domains that exhibit high affinity for PI(4,5)P2 [130, 239, 240]. In addition to Exo70p and Sec3p binding to PI(4,5)P2, the polarized concentration of PI(4,5)P2 appears to be essential to the maintenance of cell polarization, even if not critical for its establishment. Although PI(4,5)P2 diffusion appears to be reduced in regions with high densities of phosphoinositide-binding effectors, PI(4,5)P2 has limited ability to organize into membrane domains [35, 241]. The lack of intrinsic mechanisms for PI(4,5)P2 clustering presents a chicken-and-egg scenario with respect to the role PM polarization. A dynamic equilibrium of PI(4,5)P2 synthesis and phosphoinositide transfer might offer a solution to the paradox of how a freely mobile lipid is seemingly retained and localized to one membrane site. Indeed, overexpression of Osh4p increases Cdc42p localization to the PM at sites of polarization, consistent with the notion that Osh-dependent transfer of PI4P to the PM concentrates PI(4,5)P2 to maintain Cdc42p polarization [48].
In general, the phosphoinositide interaction nodes represented by Arf1p, Sec2p-Sec4p (i.e. Rab8-Rabin8), and Cdc42p might represent optimal therapeutic targets for polarized exocytosis-related disease. As one approach, structure-based design can be used to develop allosteric inhibitors and activators specific for effector PH domains. If Rabin8 shares the same lipid affinities, PI4P binding pockets are viable targets to control its dysfunction in ciliopathies and potentially in PINK-dependent Parkinson’s. More generally, phosphoinositide kinases and phosphatases, as well as phosphoinositide transfer proteins, also represent druggable factors to curb disruptions in polarized exocytosis that induce disease.
Data availability
All data of this study are available within the article.
References
Strahl T, Thorner J (2007) Synthesis and function of membrane phosphoinositides in budding yeast, Saccharomyces cerevisiae. Biochim Biophys Acta 1771(3):353–404
Sciorra VA, Audhya A, Parsons AB, Segev N, Boone C, Emr SD (2005) Synthetic genetic array analysis of the PtdIns 4-kinase Pik1p identifies components in a Golgi-specific Ypt31/rab-GTPase signaling pathway. Mol Biol Cell 16(2):776–793
Yakir-Tamang L, Gerst JE (2009) A phosphatidylinositol-transfer protein and phosphatidylinositol-4-phosphate 5-kinase control Cdc42 to regulate the actin cytoskeleton and secretory pathway in yeast. Mol Biol Cell 20(15):3583–3597
Schekman R (2010) Charting the secretory pathway in a simple eukaryote. Mol Biol Cell 21(22):3781–3784
Henry SA, Kohlwein SD, Carman GM (2012) Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics 190(2):317–349
Singh P (2017) Budding yeast: an ideal backdrop for in vivo lipid biochemistry. Front Cell Dev Biol 4:156
Guan XL, Riezman I, Wenk MR, Riezman H (2010) Yeast lipid analysis and quantification by mass spectrometry. Methods Enzymol 470:369–391
Hammond GR, Balla T (2015) Polyphosphoinositide binding domains: key to inositol lipid biology. Biochim Biophys Acta 1851(6):746–758
Ganesan S, Shabits BN, Zaremberg V (2016) Tracking diacylglycerol and phosphatidic acid pools in budding yeast. Lipid Insights 8(Suppl 1):75–85
Hammond GRV, Ricci MMC, Weckerly CC, Wills RC (2022) An update on genetically encoded lipid biosensors. Mol Biol Cell. https://doi.org/10.1091/mbc.E21-07-0363
Steinfeld N, Giridharan SSP, Kauffman EJ, Weisman LS (2021) Simultaneous detection of phosphoinositide lipids by radioactive metabolic labeling. Methods Mol Biol 2251:1–17
Audhya A, Foti M, Emr SD (2000) Distinct roles for the yeast phosphatidylinositol 4-kinases, Stt4p and Pik1p, in secretion, cell growth, and organelle membrane dynamics. Mol Biol Cell 11(8):2673–2689
Han GS, Audhya A, Markley DJ, Emr SD, Carman GM (2002) The Saccharomyces cerevisiae LSB6 gene encodes phosphatidylinositol 4-kinase activity. J Biol Chem 277(49):47709–47718
Chang FS, Han GS, Carman GM, Blumer KJ (2005) A WASp-binding type II phosphatidylinositol 4-kinase required for actin polymerization-driven endosome motility. J Cell Biol 171(1):133–142
Kourkoulou A, Martzoukou O, Fischer R, Amillis S (2024) A type II phosphatidylinositol-4-kinase coordinates sorting of cargo polarizing by endocytic recycling. Commun Biol 7(1):855
Heckle LA, Kozminski KG (2023) Osh-dependent and -independent regulation of PI4P levels during polarized growth of saccharomyces cerevisiae. Mol Biol Cell 34(11):ar104
Mizuno-Yamasaki E, Rivera-Molina F, Novick P (2012) GTPase networks in membrane traffic. Annu Rev Biochem 81:637–659
Stefan CJ, Audhya A, Emr SD (2002) The yeast synaptojanin-like proteins control the cellular distribution of phosphatidylinositol (4,5)-bisphosphate. Mol Biol Cell 13:542–557
Baird D, Stefan C, Audhya A, Weys S, Emr SD (2008) Assembly of the PtdIns 4-kinase Stt4 complex at the plasma membrane requires Ypp1 and Efr3. J Cell Biol 183(6):1061–1074
Omnus DJ, Cadou A, Thomas FB, Bader JM, Soh N, Chung GHC, Vaughan AN, Stefan CJ (2020) A heat-sensitive Osh protein controls PI4P polarity. BMC Biol 18(1):28
Foti M, Audhya A, Emr SD (2001) Sac1 lipid phosphatase and Stt4 phosphatidylinositol 4-kinase regulate a pool of phosphatidylinositol 4-phosphate that functions in the control of the actin cytoskeleton and vacuole morphology. Mol Biol Cell 12(8):2396–2411
Del Bel LM, Brill JA (2018) Sac1, a lipid phosphatase at the interface of vesicular and nonvesicular transport. Traffic 19(5):301–318
Stefan CJ, Manford AG, Baird D, Yamada-Hanff J, Mao Y, Emr SD (2011) Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 144(3):389–401
Quon E, Sere YY, Chauhan N, Johansen J, Sullivan DP, Dittman JS, Rice WJ, Chan RB, Di Paolo G, Beh CT, Menon AK (2018) Endoplasmic reticulum-plasma membrane contact sites integrate sterol and phospholipid regulation. PLoS Biol 16(5):e2003864
Nenadic A, Zaman MF, Johansen J, Volpiana MW, Beh CT (2023) Increased phospholipid flux bypasses overlapping essential requirements for the yeast Sac1p phosphoinositide phosphatase and ER-PM membrane contact sites. J Biol Chem 299(9):105092
Guo S, Stolz LE, Lemrow SM, York JD (1999) SAC1-like domains of yeast SAC1, INP52, and INP53 and of human synaptojanin encode polyphosphoinositide phosphatases. J Biol Chem 274(19):12990–12995
Garrenton LS, Stefan CJ, McMurray MA, Emr SD, Thorner J (2010) Pheromone-induced anisotropy in yeast plasma membrane phosphatidylinositol-4,5-bisphosphate distribution is required for MAPK signaling. Proc Natl Acad Sci USA 107(26):11805–11810
Wedlich-Soldner R, Altschuler S, Wu L, Li R (2003) Spontaneous cell polarization through actomyosin-based delivery of the Cdc42 GTPase. Science 299(5610):1231–1235
Desrivières S, Cooke FT, Parker PJ, Hall MN (1998) MSS4, a phosphatidylinositol-4-phosphate 5- kinase required for organization of the actin cytoskeleton in Saccharomyces cerevisiae. J Biol Chem 273(25):15787–15793
He B, Xi F, Zhang X, Zhang J, Guo W (2007) Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J 26(18):4053–4065
Zhang X, Orlando K, He B, Xi F, Zhang J, Zajac A, Guo W (2008) Membrane association and functional regulation of Sec3 by phospholipids and Cdc42. J Cell Biol 180(1):145–158
Homma K, Terui S, Minemua M, Qadota H, Anraku Y, Kanaho Y, Ohya Y (1998) Phosphatidylinositol-4-phosphate 5-kinase localized on the plasma membrane is essential for yeast cell morphogenesis. J Biol Chem 273(25):15779–15786
Yoshida S, Ohya Y, Nakano A, Anraku Y (1994) Genetic interactions among genes involved in the STT4-PKC1 pathway of Saccharomyces cerevisiae. Mol Gen Genet 242(6):631–640
Wen Y, Vogt VM, Feigenson GW (2021) PI(4,5)P2 clustering and its impact on biological functions. Annu Rev Biochem 90:681–707
Pacheco J, Cassidy AC, Zewe JP, Wills RC, Hammond GRV (2023) PI(4,5)P2 diffuses freely in the plasma membrane even within high-density effector protein complexes. J Cell Biol 222(2):e202204099
Mayinger P (2012) Phosphoinositides and vesicular membrane traffic. Biochim Biophys Acta 1821(8):1104–1113
Tan J, Oh K, Burgess J, Hipfner DR, Brill JA (2014) PI4KIIIα is required for cortical integrity and cell polarity during Drosophila oogenesis. J Cell Sci 127(5):954–966
Martin TFJ (2015) PI(4,5)P2-binding effector proteins for vesicle exocytosis. Biochim Biophys Acta 1851(6):785–793
Beh CT, McMaster CR, Kozminski KG, Menon AK (2012) A detour for yeast oxysterol binding proteins. J Biol Chem 287(14):11481–11488
Kentala H, Weber-Boyvat M, Olkkonen VM (2016) OSBP-related protein family: mediators of lipid transport and signaling at membrane contact sites. Int Rev Cell Mol Biol 321:299–340
Pietrangelo A, Ridgway ND (2018) Bridging the molecular and biological functions of the oxysterol-binding protein family. Cell Mol Life Sci 75(17):3079–3098
Beh CT, Cool L, Phillips J, Rine J (2001) Overlapping functions of the yeast oxysterol-binding protein homologues. Genetics 157(3):1117–1140
Ridgway ND, Dawson PA, Brown MS, Goldstein JL (1992) Translocation of oxysterol binding protein to Golgi apparatus triggered by ligand binding. J Cell Biol 116(2):307–319
de Saint-Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B, Drin D (2011) Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J Cell Biol 195(6):965–978
Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P (2015) PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 349(6246):428–432
Sohn M, Korzeniowski M, Zewe JP, Wills RC, Hammond GRV, Humpolickova J, Vrzal L, Chalupska D, Veverka V, Fairn GD, Boura E, Balla T (2018) PI(4,5)P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER–PM contact sites. J Cell Biol 217:1797–1813
LeBlanc MA, McMaster CR (2010) Surprising roles for phospholipid binding proteins revealed by high throughput genetics. Biochem Cell Biol 88(4):565–574
Kozminski KG, Alfaro G, Dighe S, Beh CT (2006) Homologues of oxysterol-binding proteins affect Cdc42p- and Rho1p-mediated cell polarization in Saccharomyces cerevisiae. Traffic 7(9):1224–1242
Alfaro G, Johansen J, Dighe SA, Duamel G, Kozminski KG, Beh CT (2011) The sterol-binding protein Kes1/Osh4p is a regulator of polarized exocytosis. Traffic 12(11):1521–1536
Ling Y, Hayano S, Novick P (2014) Osh4p is needed to reduce the level of phosphatidylinositol-4- phosphate on secretory vesicles as they mature. Mol Biol Cell 25:3389–3400
Smindak RJ, Heckle LA, Chittari SS, Hand MA, Hyatt DM, Mantus GE, Sanfelippo WA, Kozminski KG (2017) Lipid-dependent regulation of exocytosis in S. cerevisiae by OSBP homolog (Osh) 4. J Cell Sci 130(22):3891–3906
Aitken JF, van Heusden GP, Temkin M, Dowhan W (1990) The gene encoding the phosphatidylinositol transfer protein is essential for cell growth. J Biol Chem 265(8):4711–4717
Hama H, Schnieders EA, Thorner J, Takemoto JY, DeWald DB (1999) Direct involvement of phosphatidylinositol 4-phosphate in secretion in the yeast Saccharomyces cerevisiae. J Biol Chem 274(48):34294–34300
Curwin AJ, Fairn GD, McMaster CR (2009) Phospholipid transfer protein Sec14 is required for trafficking from endosomes and regulates distinct trans-Golgi export pathways. J Biol Chem 284(11):7364–7375
Novick P, Schekman R (1979) Secretion and cell-surface growth are blocked in a temperaturesensitive mutant of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 76(4):1858–1862
Nishimura T, Gecht M, Covino R, Hummer G, Surma MA, Klose C, Arai H, Kono N, Stefan CJ (2019) Osh proteins control nanoscale lipid organization necessary for PI(4,5)P2 synthesis. Mol Cell 75(5):1043–1057
Novick P, Field C, Schekman R (1980) Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 21(1):205–215
Bankaitis VA, Aitken JR, Cleves AE, Dowhan W (1990) An essential role for a phospholipid transfer protein in yeast Golgi function. Nature 347(6293):561–562
Phillips SE, Sha B, Topalof L, Xie Z, Alb JG, Klenchin VA, Swigart P, Cockcroft S, Martin TF, Luo M, Bankaitis VA (1999) Yeast Sec14p deficient in phosphatidylinositol transfer activity is functional in vivo. Mol Cell 4(2):187–197
Tahotna D, Holic R, Poloncova K, Simockova M, Griac P (2007) Phosphatidylcholine transfer activity of yeast Sec14p is not essential for its function in vivo. Biochim Biophys Acta 1771(1):83–92
Cleves AE, McGee TP, Whitters EA, Champion KM, Aitken JR, Dowhan W, Goebl M, Bankaitis VA (1991) Mutations in the CDP-choline pathway for phospholipid biosynthesis bypass the requirement for an essential phospholipid transfer protein. Cell 64(4):789–800
Fang M, Kearns BG, Gedvilaite A, Kagiwada S, Kearns M, Fung MK, Bankaitis VA (1996) Kes1p shares homology with human oxysterol binding protein and participates in a novel regulatory pathway for yeast Golgi-derived transport vesicle biogenesis. EMBO J 15(23):6447–6459
Fairn GD, Curwin AJ, Stefan CJ, McMaster CR (2007) The oxysterol binding protein Kes1p regulates Golgi apparatus phosphatidylinositol-4-phosphate function. Proc Natl Acad Sci USA 104(39):15352–15357
Rivas MP, Kearns BG, Xie Z, Guo S, Sekar MC, Hosaka K, Kagiwada S, York JD, Bankaitis VA (1999) Pleiotropic alterations in lipid metabolism in yeast sac1 mutants: relationship to “bypass Sec14p” and inositol auxotrophy. Mol Biol Cell 10(7):2235–2250
Xie Z, Fang M, Rivas MP, Faulkner AJ, Sternweis PC, Engebrecht JA, Bankaitis VA (1998) Phospholipase D activity is required for suppression of yeast phosphatidylinositol transfer protein defects. Proc Natl Acad Sci USA 95(21):12346–12351
Johansen J, Ramanathan V, Beh CT (2012) Vesicle trafficking from a lipid perspective: lipid regulation of exocytosis in Saccharomyces cerevisiae. Cellular Logist 2(3):151–160
Skinner HB, McGee TP, McMaster CR, Fry MR, Bell RM, Bankaitis VA (1995) The Saccharomyces cerevisiae phosphatidylinositol-transfer protein effects a ligand-dependent inhibition of choline-phosphate cytidylyltransferase activity. Proc Natl Acad Sci USA 92(1):112–116
Allan D, Thomas P, Michell RH (1978) Rapid transbilayer diffusion of 1,2-diacylglycerol and its relevance to control of membrane curvature. Nature 276(5685):289–290
Asp L, Kartberg F, Fernandez-Rodriguez J, Smedh M, Elsner M, Laporte F, Bárcena M, Jansen KA, Valentijn JA, Koster AJ, Bergeron JJ, Nilsson T (2009) Early stages of Golgi vesicle and tubule formation require diacylglycerol. Mol Biol Cell 20(3):780–790
Shemesh T, Luini A, Malhotra V, Burger KN, Kozlov MM (2003) Prefission constriction of Golgi tubular carriers driven by local lipid metabolism: a theoretical model. Biophys J 85(6):3813–3827
Campelo F, van Galen J, Turacchio G, Parashuraman S, Kozlov MM, García-Parajo MF, Malhotra V (2017) Sphingomyelin metabolism controls the shape and function of the Golgi cisternae. eLife 6:e24603
Gloor Y, Schöne M, Habermann B, Ercan E, Beck M, Weselek G, Müller-Reichert T, WalchSolimena C (2010) Interaction between Sec7p and Pik1p: the first clue for the regulation of a coincidence detection signal. Eur J Cell Biol 89(8):575–583
Thomas LL, Highland CM, Fromme JC (2021) Arf1 orchestrates Rab GTPase conversion at the trans-Golgi network. Mol Biol Cell 32(11):1104–1120
Richardson BC, McDonold CM, Fromme JC (2012) The Sec7 Arf-GEF is recruited to the transGolgi network by positive feedback. Dev Cell 22(4):799–810
McDonold CM, Fromme JC (2014) Four GTPases differentially regulate the Sec7 Arf-GEF to direct traffic at the trans-golgi network. Dev Cell 30(6):759–767
Lipatova Z, Segev N (2019) Ypt/Rab GTPases and their TRAPP GEFs at the Golgi. FEBS Lett 593(17):2488–2500
Highland CM, Fromme JC (2021) Arf1 directly recruits the Pik1-Frq1 PI4K complex to regulate the final stages of Golgi maturation. Mol Biol Cell 32(10):1064–1080
Poon PP, Nothwehr SF, Singer RA, Johnston GC (2001) The Gcs1 and Age2 ArfGAP proteins provide overlapping essential function for transport from the yeast trans-Golgi network. J Cell Biol 155(7):1239–1250
Yanagisawa LL, Marchena J, Xie Z, Li X, Poon PP, Singer RA, Johnston GC, Randazzo PA, Bankaitis VA (2002) Activity of specific lipid-regulated ADP ribosylation factor-GTPase-activating proteins is required for Sec14p-dependent Golgi secretory function in yeast. Mol Biol Cell 13(7):2193–2206
Wood CS, Hung CS, Huoh YS, Mousley CJ, Stefan CJ, Bankaitis V, Ferguson KM, Burd CG (2012) Local control of phosphatidylinositol 4-phosphate signaling in the Golgi apparatus by Vps74 and Sac1 phosphoinositide phosphatase. Mol Biol Cell 23(13):2527–2536
Tahirovic S, Schorr M, Mayinger P (2005) Regulation of intracellular phosphatidylinositol-4- phosphate by the Sac1 lipid phosphatase. Traffic 6:116–130
Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B (2013) A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155(4):830–843
Doyle CP, Timple L, Hammond GRV (2024) OSBP is a Major Determinant of Golgi Phosphatidylinositol 4-Phosphate Homeostasis. Contact (Thousand Oaks) 7:25152564241232196
Tóth B, Balla A, Ma H, Knight ZA, Shokat KM, Balla T (2006) Phosphatidylinositol 4-kinase IIIbeta regulates the transport of ceramide between the endoplasmic reticulum and Golgi. J Biol Chem 281(47):36369–36377
Perry RJ, Ridgway ND (2006) Oxysterol-binding protein and vesicle-associated membrane protein-associated protein are required for sterol-dependent activation of the ceramide transport protein. Mol Biol Cell 17(6):2604–2616
Wyles JP, McMaster CR, Ridgway ND (2002) Vesicle-associated membrane protein-associated protein-A (VAP-A) interacts with the oxysterol-binding protein to modify export from the endoplasmic reticulum. J Biol Chem 277(33):29908–29918
Toulmay A, Whittle FB, Yang J, Bai X, Diarra J, Banerjee S, Levine TP, Golden A, Prinz WA (2022) Vps13-like proteins provide phosphatidylethanolamine for GPI anchor synthesis in the ER. J Cell Biol 221(3):e202111095
Natarajan P, Liu K, Patil DV, Sciorra VA, Jackson CL, Graham TR (2009) Regulation of a Golgi flippase by phosphoinositides and an ArfGEF. Nat Cell Biol 11(12):1421–1426
Hankins HM, Sere YY, Diab NS, Menon AK, Graham TR (2015) Phosphatidylserine translocation at the yeast trans-Golgi network regulates protein sorting into exocytic vesicles. Mol Biol Cell 26(25):4674–4685
Jacquot A, Montigny C, Hennrich H, Barry R, le Maire M, Jaxel C, Holthuis J, Champeil P, Lenoir G (2012) Phosphatidylserine stimulation of Drs2p·Cdc50p lipid translocase dephosphorylation is controlled by phosphatidylinositol-4-phosphate. J Biol Chem 287(16):13249–13261
Zhou X, Sebastian TT, Graham TR (2013) Auto-inhibition of Drs2p, a yeast phospholipid flippase, by its carboxyl-terminal tail. J Biol Chem 288(44):31807–31815
Gall WE, Geething NC, Hua Z, Ingram MF, Liu K, Chen SI, Graham TR (2002) Drs2p-dependent formation of exocytic clathrin-coated vesicles in vivo. Curr Biol 12(18):1623–1627
Alder-Baerens N, Lisman Q, Luong L, Pomorski T, Holthuis JC (2006) Loss of P4 ATPases Drs2p and Dnf3p disrupts aminophospholipid transport and asymmetry in yeast post-Golgi secretory vesicles. Mol Biol Cell 17(4):1632–1642
Liu K, Surendhran K, Nothwehr SF, Graham TR (2008) P4-ATPase requirement for AP-1/clathrin function in protein transport from the trans-Golgi network and early endosomes. Mol Biol Cell 19(8):3526–3535
Xu P, Baldridge RD, Chi RJ, Burd CG, Graham TR (2013) Phosphatidylserine flipping enhances membrane curvature and negative charge required for vesicular transport. J Cell Biol 202(6):875–886
Muthusamy BP, Raychaudhuri S, Natarajan P, Abe F, Liu K, Prinz WA, Graham TR (2009) Control of protein and sterol trafficking by antagonistic activities of a type IV P-type ATPase and oxysterol binding protein homologue. Mol Biol Cell 20(12):2920–2931
Surma MA, Klose C, Klemm RW, Ejsing CS, Simons K (2011) Generic sorting of raft lipids into secretory vesicles in yeast. Traffic 12(9):1139–1147
Wang CW, Hamamoto S, Orci L, Schekman R (2006) Exomer: a coat complex for transport of select membrane proteins from the trans-Golgi network to the plasma membrane in yeast. J Cell Biol 174(7):973–983
Anton-Plagaro C, Sanchez N, Valle R, Mulet JM, Duncan MC, Roncero C (2021) Exomer complex regulates protein traffic at the TGN through differential interactions with cargos and clathrin adaptor complexes. FASEB J 35(6):e21615
Harsay E, Bretscher A (1995) Parallel secretory pathways to the cell surface in yeast. J Cell Biol 131(2):297–310
Papanikou E, Glick BS (2014) Golgi compartmentation and identity. Curr Opin Cell Biol 29:74–81
Graham TR, Burd CG (2011) Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol 21(2):113–121
Novick P (2016) Regulation of membrane traffic by Rab GEF and GAP cascades. Small GTPases 7(4):252–256
Thomas LL, Fromme JC (2016) GTPase cross talk regulates TRAPPII activation of Rab11 homologues during vesicle biogenesis. J Cell Biol 215(4):499–513
Jedd G, Mulholland J, Segev N (1997) Two new Ypt GTPases are required for exit from the yeast trans-Golgi compartment. J Cell Biol 137(3):563–580
Walch-Solimena C, Collins RN, Novick PJ (1997) Sec2p mediates nucleotide exchange on Sec4p and is involved in polarized delivery of post-Golgi vesicles. J Cell Biol 137(7):1495–1509
Ortiz D, Medkova M, Walch-Solimena C, Novick P (2002) Ypt32 recruits the Sec4p guanine nucleotide exchange factor, Sec2p, to secretory vesicles; evidence for a Rab cascade in yeast. J Cell Biol 157(6):1005–1015
Mizuno-Yamasaki E, Medkova M, Coleman J, Novick P (2010) Phosphatidylinositol 4-phosphate controls both membrane recruitment and a regulatory switch of the Rab GEF Sec2p. Dev Cell 18(5):828–840
Lipatova Z, Tokarev AA, Jin Y, Mulholland J, Weisman LS, Segev N (2008) Direct interaction between a myosin V motor and the Rab GTPases Ypt31/32 is required for polarized secretion. Mol Biol Cell 19(10):4177–4187
Santiago-Tirado FH, Legesse-Miller A, Schott D, Bretscher A (2011) PI4P and Rab inputs collaborate in myosin-V-dependent transport of secretory compartments in yeast. Dev Cell 20(1):47–59
Jin Y, Sultana A, Gandhi P, Franklin E, Hamamoto S, Khan AR, Munson M, Schekman R, Weisman LS (2011) Myosin V transports secretory vesicles via a Rab GTPase cascade and interaction with the exocyst complex. Dev Cell 21(6):1156–1170
Makowski SL, Kuna RS, Field SJ (2020) Induction of membrane curvature by proteins involved in Golgi trafficking. Adv Biol Regul 75:100661
Gingras RM, Sulpizio AM, Park J, Bretscher A (2022) High-resolution secretory timeline from vesicle formation at the Golgi to fusion at the plasma membrane in S. cerevisiae. eLife 11:e78750
Hsu SC, TerBush D, Abraham M, Guo W (2004) The exocyst complex in polarized exocytosis. Int Rev Cytol 233:243–265
Sato Y, Shirakawa R, Horiuchi H, Dohmae N, Fukai S, Nureki O (2007) Asymmetric coiled-coil structure with Guanine nucleotide exchange activity. Structure 15(2):245–252
Mei K, Guo W (2018) The exocyst complex. Curr Biol 28(17):R922–R925
Stalder D, Novick PJ (2016) The casein kinases Yck1p and Yck2p act in the secretory pathway, in part, by regulating the Rab exchange factor Sec2p. Mol Biol Cell 27(4):686–701
Miller BK, Rossi G, Hudson S, Cully D, Baker RW, Brennwald P (2023) Allosteric regulation of exocyst: discrete activation of tethering by two spatial signals. J Cell Biol 222(3):e202206108
Costanzo M, VanderSluis B, Koch EN et al (2016) A global genetic interaction network maps a wiring diagram of cellular function. Science 353(6306):aaf1420
Beh CT, Rine J (2004) A role for yeast oxysterol-binding protein homologs in endocytosis and in the maintenance of intracellular sterol-lipid distribution. J Cell Sci 117(Pt 14):2983–2996
Routt SM, Ryan MM, Tyeryar K, Rizzieri KE, Mousley C, Roumanie O, Brennwald PJ, Bankaitis VA (2005) Nonclassical PITPs activate PLD via the Stt4p PtdIns-4-kinase and modulate function of late stages of exocytosis in vegetative yeast. Traffic 6(12):1157–1172
Masters TA, Sheetz MP, Gauthier NC (2016) F-actin waves, actin cortex disassembly and focal exocytosis driven by actin-phosphoinositide positive feedback. Cytoskeleton 73(4):180–196
Govindan B, Novick P (1995) Development of cell polarity in budding yeast. J Exp Zool 273(5):401–424
Pruyne D, Bretscher A (2000) Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. J Cell Sci 113(3):365–375
Pruyne DW, Schott DH, Bretscher A (1998) Tropomyosin-containing actin cables direct the Myo2p-dependent polarized delivery of secretory vesicles in budding yeast. J Cell Biol 143(7):1931–1945
Etienne-Manneville S (2004) Cdc42–the centre of polarity. J Cell Sci 117(Pt 8):1291–1300
Wedlich-Soldner R, Wai SC, Schmidt T, Li R (2004) Robust cell polarity is a dynamic state established by coupling transport and GTPase signaling. J Cell Biol 166(6):889–900
Meca J, Massoni-Laporte A, Martinez D, Sartorel E, Loquet A, Habenstein B, McCusker D (2019) Avidity-driven polarity establishment via multivalent lipid-GTPase module interactions. EMBO J 38(3):e99652
Toenjes KA, Sawyer MM, Johnson DI (1999) The guanine-nucleotide-exchange factor Cdc24p is targeted to the nucleus and polarized growth sites. Curr Biol 9(20):1183–1186
Orlando K, Zhang J, Zhang X, Yue P, Chiang T, Bi E, Guo W (2008) Regulation of Gic2 localization and function by phosphatidylinositol 4,5-bisphosphate during the establishment of cell polarity in budding yeast. J Biol Chem 283(21):14205–14212
Liu Y, Zhang T, Sun D, Luo G (2019) The Cdc42 effectors Gic1 and Gic2 regulate polarized postGolgi secretion. Cell Biosci 9:33
Adamo JE, Rossi G, Brennwald P (1999) The Rho GTPase Rho3 has a direct role in exocytosis that is distinct from its role in actin polarity. Mol Biol Cell 10(12):4121–4133
Robinson NG, Guo L, Imai J, Toh-E A, Matsui Y, Tamanoi F (1999) Rho3 of Saccharomyces cerevisiae, which regulates the actin cytoskeleton and exocytosis, is a GTPase which interacts with Myo2 and Exo70. Mol Cell Biol 19(5):3580–3587
Liu D, Novick P (2014) Bem1p contributes to secretory pathway polarization through a direct interaction with Exo70p. J Cell Biol 207(1):59–72
Pleskot R, Cwiklik L, Jungwirth P, Žárský V (1848) Potocký M (2015) Membrane targeting of the yeast exocyst complex. Biochim Biophys Acta 7:1481–1489
Helliwell SB, Schmidt A, Ohya Y, Hall MN (1998) The Rho1 effector Pkc1, but not Bni1, mediates signalling from Tor2 to the actin cytoskeleton. Curr Biol 8(22):1211–1214
Guo W, Tamanoi F, Novick P (2001) Spatial regulation of the exocyst complex by Rho1 GTPase. Nat Cell Biol 3(4):353–360
Zhang X, Bi E, Novick P, Du L, Kozminski K, Lipschutz J, Guo W (2001) Cdc42 interacts with the exocyst and regulates polarized secretion. J Biol Chem 276(50):46745–46750
Zhang X, Wang P, Gangar A, Zhang J, Brennwald P, TerBush D, Guo W (2005) Lethal giant larvae proteins interact with the exocyst complex and are involved in polarized exocytosis. J Cell Biol 170(2):273–283
Zhang X, Zajac A, Zhang J, Wang P, Li M, Murray J, TerBush GW (2005) The critical role of Exo84p in the organization and polarized localization of the exocyst complex. J Biol Chem 280(21):20356–20364
Wu H, Turner C, Gardner J, Temple B, Brennwald P (2010) The Exo70 subunit of the exocyst is an effector for both Cdc42 and Rho3 function in polarized exocytosis. Mol Biol Cell 21(3):430–442
Rossi G, Lepore D, Kenner L, Czuchra A, Plooster M, Frost A, Munson M, Brennwald P (2020) Exocyst structural changes associated with activation of tethering downstream of Rho/Cdc42 GTPases. J Cell Biol 219(2):e201904161
Yamashita M, Kurokawa K, Sato Y, Yamagata A, Mimura H, Yoshikawa A, Sato K, Nakano A, Fukai S (2010) Structural basis for the Rho- and phosphoinositide-dependent localization of the exocyst subunit Sec3. Nat Struct Mol Biol 17(2):180–186
Boyd C, Hughes T, Pypaert M, Novick P (2004) Vesicles carry most exocyst subunits to exocytic sites marked by the remaining two subunits, Sec3p and Exo70p. J Cell Biol 167(5):889–901
Jin R, Junutula JR, Matern HT, Ervin KE, Scheller RH, Brunger AT (2005) Exo84 and Sec5 are competitive regulatory Sec6/8 effectors to the RalA GTPase. EMBO J 24(12):2064–2074
Moskalenko S, Tong C, Rosse C, Mirey G, Formstecher E, Daviet L, Camonis J, White MA (2003) Ral GTPases regulate exocyst assembly through dual subunit interactions. J Biol Chem 278(51):51743–51748
Nishida-Fukuda H (2019) The exocyst: dynamic machine or static tethering complex? BioEssays 41(8):e1900056
Maib H, Murray DH (2022) A mechanism for exocyst-mediated tethering via Arf6 and PIP5K1Cdriven phosphoinositide conversion. Curr Biol 32(13):2821-2833.e6
Heider MR, Gu M, Duffy CM, Mirza AM, Marcotte LL, Walls AC, Farrall N, Hakhverdyan Z, Field MC, Rout MP, Frost A, Munson M (2016) Subunit connectivity, assembly determinants and architecture of the yeast exocyst complex. Nat Struct Mol Biol 23(1):59–66
Picco A, Irastorza-Azcarate I, Specht T, Böke D, Pazos I, Rivier-Cordey AS, Devos DP, Kaksonen M, Gallego O (2017) The in vivo architecture of the exocyst provides structural basis for exocytosis. Cell 168(3):400-412.e18
TerBush DR, Maurice T, Roth D, Novick P (1996) The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J 15(23):6483–6494
Munson M, Novick P (2006) The exocyst defrocked, a framework of rods revealed. Nat Struct Mol Biol 13(7):577–581
Shen D, Yuan H, Hutagalung A, Verma A, Kümmel D, Wu X, Reinisch K, McNew J, Novick P (2013) The synaptobrevin homologue Snc2p recruits the exocyst to secretory vesicles by binding to Sec6p. J Cell Biol 202(3):509–526
Dubuke ML, Maniatis S, Shaffer SA, Munson M (2015) The exocyst subunit Sec6 interacts with assembled exocytic SNARE complexes. J Biol Chem 290(47):28245–28256
Yue P, Zhang Y, Mei K, Wang S, Lesigang J, Zhu Y, Dong G, Guo W (2017) Sec3 promotes the initial binary t-SNARE complex assembly and membrane fusion. Nat Commun 8:14236
Lehman K, Rossi G, Adamo JE, Brennwald P (1999) Yeast homologues of tomosyn and lethal giant larvae function in exocytosis and are associated with the plasma membrane SNARE, Sec9. J Cell Biol 146(1):125–140
Grosshans BL, Andreeva A, Gangar A, Niessen S, Yates JR 3rd, Brennwald P, Novick P (2006) The yeast lgl family member Sro7p is an effector of the secretory Rab GTPase Sec4p. J Cell Biol 172(1):55–66
Rossi G, Watson K, Kennedy W, Brennwald P (2018) The tomosyn homologue, Sro7, is a direct effector of the Rab GTPase, Sec4, in post-Golgi vesicle tethering. Mol Biol Cell 29(12):1476–1486
Coluccio A, Malzone M, Neiman AM (2004) Genetic evidence of a role for membrane lipid composition in the regulation of soluble NEM-sensitive factor receptor function in Saccharomyces cerevisiae. Genetics 166(1):89–97
Mendonsa R, Engebrecht J (2009) Phosphatidylinositol-4,5-bisphosphate and phospholipase dgenerated phosphatidic acid specify SNARE-mediated vesicle fusion for prospore membrane formation. Eukaryot Cell 8(8):1094–1105
Min K, Neiman AM, Konopka JB (2020) Fungal pathogens: shape-shifting invaders. Trends Microbiol 28(11):922–933
Ghugtyal V, Garcia-Rodas R, Seminara A, Schaub S, Bassilana M, Arkowitz RA (2015) Phosphatidylinositol-4-phosphate-dependent membrane traffic is critical for fungal filamentous growth. Proc Natl Acad Sci U S A 112(28):8644–8649
Garcia-Rodas R, Labbaoui H, Orange F, Solis N, Zaragoza O, Filler SG, Bassilana M, Arkowitz RA (2021) Plasma membrane phosphatidylinositol-4-phosphate is not necessary for Candida albicans viability yet Is key for cell wall integrity and systemic infection. mBio 13(1):e0387321
Konopka JB (2022) Plasma membrane phosphatidylinositol 4-phosphate Is necessary for virulence of Candida albicans. mBio 13(3):e0036622
Vernay A, Schaub S, Guillas I, Bassilana M, Arkowitz RA (2012) A steep phosphoinositide bis-phosphate gradient forms during fungal filamentous growth. J Cell Biol 198(4):711–730
Lanze CE, Konopka JB (2024) Sur7 mediates a novel pathway for PI4,5P2 regulation in C. albicans that promotes stress resistance and cell wall morphogenesis. Mol Biol Cell 35(7):99
Burgett AW, Poulsen TB, Wangkanont K et al (2011) Natural products reveal cancer cell dependence on oxysterol-binding proteins. Nat Chem Biol 7(9):639–647. https://doi.org/10.1038/nchembio.625
Mähs A, Ischebeck T, Heilig Y, Stenzel I, Hempel F, Seiler S, Heilmann I (2012) The essential phosphoinositide kinase MSS-4 is required for polar hyphal morphogenesis, localizing to sites of growth and cell fusion in Neurospora crassa. PLoS ONE 7(12):e51454
Takeshita N, Evangelinos M, Zhou L, Serizawa T, Somera-Fajardo RA, Lu L, Takaya N, Nienhaus GU, Fischer R (2017) Pulses of Ca2+ coordinate actin assembly and exocytosis for stepwise cell extension. Proc Natl Acad Sci U S A 114(22):5701–5706
Fukuda S, Yamamoto R, Yanagisawa N, Takaya N, Sato Y, Riquelme M, Takeshita N (2021) Trade-off between plasticity and velocity in mycelial growth. mBio 12(2):e03196-20
Lopez MC, Nicaud JM, Skinner HB, Vergnolle C, Kader JC, Bankaitis VA, Gaillardin C (1994) A phosphatidylinositol/phosphatidylcholine transfer protein is required for differentiation of the dimorphic yeast Yarrowia lipolytica from the yeast to the mycelial form. J Cell Biol 125(1):113–127
Riggle PJ, Slobodkin IV, Brown DH, Hanson MP, Volkert TL, Kumamoto CA (1997) Two transcripts, differing at their 3’ ends, are produced from the Candida albicans SEC14 gene. Microbiology 143(Pt 11):3527–3535
Chayakulkeeree M, Johnston SA, Oei JB, Lev S, Williamson PR, Wilson CF, Zuo X, Leal AL, Vainstein MH, Meyer W, Sorrell TC, May RC, Djordjevic JT (2011) SEC14 is a specific requirement for secretion of phospholipase B1 and pathogenicity of Cryptococcus neoformans. Mol Microbiol 80(4):1088–1101
Khan D, McGrath KR, Dorosheva O, Bankaitis VA, Tripathi A (2016) Structural elements that govern Sec14-like PITP sensitivities to potent small molecule inhibitors. J Lipid Res 57(4):650–662
Khan D, Nile AH, Tripathi A, Bankaitis VA (2021) Emerging prospects for combating fungal infections by targeting phosphatidylinositol transfer proteins. Int J Mol Sci 22(13):6754
Köhli M, Galati V, Boudier K, Roberson RW, Philippsen P (2008) Growth-speed-correlated localization of exocyst and polarisome components in growth zones of Ashbya gossypii hyphal tips. J Cell Sci 121(Pt 23):3878–3889
Taheri-Talesh N, Horio T, Araujo-Bazán L, Dou X, Espeso EA, Peñalva MA, Osmani SA, Oakley BR (2008) The tip growth apparatus of Aspergillus nidulans. Mol Biol Cell 19(4):1439–1449
Richthammer C, Enseleit M, Sanchez-Leon E, März S, Heilig Y, Riquelme M, Seiler S (2012) RHO1 and RHO2 share partially overlapping functions in the regulation of cell wall integrity and hyphal polarity in Neurospora crassa. Mol Microbiol 85(4):716–733
Chen X, Ebbole DJ, Wang Z (2015) The exocyst complex: delivery hub for morphogenesis and pathogenesis in filamentous fungi. Curr Opin Plant Biol 28:48–54
Douglas LM, Konopka JB (2016) Plasma membrane organization promotes virulence of the human fungal pathogen Candida albicans. J Microbiol 54(3):178–191
Jones LA, Sudbery PE (2010) Spitzenkorper, exocyst, and polarisome components in Candida albicans hyphae show different patterns of localization and have distinct dynamic properties. Eukaryot Cell 9(10):1455–1465
Riquelme M, Sánchez-León E (2014) The Spitzenkörper: a choreographer of fungal growth and morphogenesis. Curr Opin Microbiol 20:27–33
Silva PM, Puerner C, Seminara A, Bassilana M, Arkowitz RA (2019) Secretory vesicle clustering in fungal filamentous cells does not require directional growth. Cell Rep 28(8):2231-2245.e5
Riquelme M, Bredeweg EL, Callejas-Negrete O, Roberson RW, Ludwig S, Beltrán-Aguilar A, Seiler S, Novick P, Freitag M (2014) The Neurospora crassa exocyst complex tethers Spitzenkörper vesicles to the apical plasma membrane during polarized growth. Mol Biol Cell 25(8):1312–1326
Li CR, Lee RT, Wang YM, Zheng XD, Wang Y (2007) Candida albicans hyphal morphogenesis occurs in Sec3p-independent and Sec3p-dependent phases separated by septin ring formation. J Cell Sci 120(Pt 11):1898–1907
Bishop A, Lane R, Beniston R, Chapa-y-Lazo B, Smythe C, Sudbery P (2010) Hyphal growth in Candida albicans requires the phosphorylation of Sec2 by the Cdc28-Ccn1/Hgc1 kinase. EMBO J 29(17):2930–2942
Court H, Sudbery P (2007) Regulation of Cdc42 GTPase activity in the formation of hyphae in Candida albicans. Mol Biol Cell 18(1):265–281
Kwon MJ, Arentshorst M, Roos ED, van den Hondel CA, Meyer V, Ram AF (2011) Functional characterization of Rho GTPases in Aspergillus niger uncovers conserved and diverged roles of Rho proteins within filamentous fungi. Mol Microbiol 79(5):1151–1167
Dünkler A, Wendland J (2007) Candida albicans Rho-type GTPase-encoding genes required for polarized cell growth and cell separation. Eukaryot Cell 6(5):844–854
Corvest V, Bogliolo S, Follette P, Arkowitz RA, Bassilana M (2013) Spatiotemporal regulation of Rho1 and Cdc42 activity during Candida albicans filamentous growth. Mol Microbiol 89(4):626–648
Arkowitz RA, Bassilana M (2014) Rho GTPase-phosphatidylinositol phosphate interplay in fungal cell polarity. Biochem Soc Trans 42(1):206–211. https://doi.org/10.1042/BST20130226
Volpatti JR, Al-Maawali A, Smith L, Al-Hashim A, Brill JA, Dowling JJ (2019) The expanding spectrum of neurological disorders of phosphoinositide metabolism. Dis Model Mech 12(8):dmm038174
Derkaczew M, Martyniuk P, Hofman R, Rutkowski K, Osowski A, Wojtkiewicz J (2023) The genetic background of abnormalities in metabolic pathways of phosphoinositides and their linkage with the myotubular myopathies, neurodegenerative disorders, and carcinogenesis. Biomolecules 13(10):1550
Tokuda E, Itoh T, Hasegawa J, Ijuin T, Takeuchi Y, Irino Y, Fukumoto M, Takenawa T (2014) Phosphatidylinositol 4-phosphate in the Golgi apparatus regulates cell-cell adhesion and invasive cell migration in human breast cancer. Cancer Res 74(11):3054–3066
Billcliff PG, Lowe M (2014) Inositol lipid phosphatases in membrane trafficking and human disease. Biochem J 461(2):159–175
Cao M, Wu Y, Ashrafi G, McCartney AJ, Wheeler H, Bushong EA, Boassa D, Ellisman MH, Ryan TA, De Camilli P (2017) Parkinson Sac domain mutation in Synaptojanin 1 impairs clathrin uncoating at synapses and triggers dystrophic changes in dopaminergic axons. Neuron 93(4):882-896.e5
Rafiq MN, Fujise K, Rosenfeld MS, Xu P, De Camilli P (2024) Parkinsonism Sac domain mutation in Synaptojanin-1 affects ciliary properties in iPSC-derived dopaminergic neurons. Proc Natl Acad Sci USA 121(17):e2318943121
Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA, McInnes RR, Nussbaum RL (1992) The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature 358(6383):239–242
Nakatsu F, Messa M, Nández R, Czapla H, Zou Y, Strittmatter SM, De Camilli P (2015) Sac2/INPP5F is an inositol 4-phosphatase that functions in the endocytic pathway. J Cell Biol 209(1):85–95
Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan TA, De Camilli P (2004) Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431(7007):415–422
D’Angelo G, Vicinanza M, Di Campli A, De Matteis MA (2008) The multiple roles of PtdIns(4)P—not just the precursor of PtdIns(4,5)P2. J Cell Sci 121(Pt 12):1955–1963
Wen PJ, Osborne SL, Meunier FA (2011) Dynamic control of neuroexocytosis by phosphoinositides in health and disease. Prog Lipid Res 50(1):52–61
Wen PJ, Osborne SL, Meunier FA (2012) Phosphoinositides in neuroexocytosis and neuronal diseases. Curr Top Microbiol Immunol 362:87–98
Schwab SG, Knapp M, Sklar P, Eckstein GN, Sewekow C, Borrmann-Hassenbach M, Albus M, Becker T, Hallmayer JF, Lerer B, Maier W, Wildenauer DB (2006) Evidence for association of DNA sequence variants in the phosphatidylinositol-4-phosphate 5-kinase II alpha gene (PIP5K2A) with schizophrenia. Mol Psychiatry 11(9):837–846
Narkis G, Ofir R, Landau D, Manor E, Volokita M, Hershkowitz R, Elbedour K, Birk OS (2007) Lethal contractural syndrome type 3 (LCCS3) is caused by a mutation in PIP5K1C, which encodes PIPKI gamma of the phophatidylinsitol pathway. Am J Hum Genet 81(3):530–539
Wang Y, Lian L, Golden JA, Morrisey EE, Abrams CS (2007) PIP5KI gamma is required for cardiovascular and neuronal development. Proc Natl Acad Sci USA 104(28):11748–11753
Salter CG, Cai Y, Lo B, Helman G, Taylor H, McCartney A, Leslie JS, Accogli A, Zara F, Traverso M, Fasham J, Lees JA, Ferla MP, Chioza BA, Wenger O, Scott E, Cross HE, Crawford J, Warshawsky I, Keisling M, Baple EL (2021) Biallelic PI4KA variants cause neurological, intestinal and immunological disease. Brain 144(12):3597–3610
Verdura E, Rodríguez-Palmero A, Vélez-Santamaria V, Planas-Serra L, de la Calle I, RaspallChaure M, Roubertie A, Benkirane M, Saettini F, Pavinato L, Mandrile G, O’Leary M, O’Heir E, Barredo E, Chacón A, Michaud V, Goizet C, Ruiz M, Schlüter A, Rouvet I, Pujol A (2021) Biallelic PI4KA variants cause a novel neurodevelopmental syndrome with hypomyelinating leukodystrophy. Brain 144(9):2659–2669
Burke JE, Triscott J, Emerling BM, Hammond GRV (2023) Beyond PI3Ks: targeting phosphoinositide kinases in disease. Nat Rev Drug Discov 22(5):357–386
Das A, Guo W (2011) Rabs and the exocyst in ciliogenesis, tubulogenesis and beyond. Trends Cell Biol 21(7):383–386
Martin-Urdiroz M, Deeks MJ, Horton CG, Dawe HR, Jourdain I (2016) The exocyst complex in health and disease. Front Cell Dev Biol 4:24
Halim DO, Munson M, Gao FB (2023) The exocyst complex in neurological disorders. Hum Genet 142(8):1263–1270
Pereira C, Stalder D, Anderson GSF, Shun-Shion AS, Houghton J, Antrobus R, Chapman MA, Fazakerley DJ, Gershlick DC (2023) The exocyst complex is an essential component of the mammalian constitutive secretory pathway. J Cell Biol 222(5):e202205137
Das A, Gajendra S, Falenta K, Oudin MJ, Peschard P, Feng S, Wu B, Marshall CJ, Doherty P, Guo W, Lalli G (2014) RalA promotes a direct exocyst-Par6 interaction to regulate polarity in neuronal development. J Cell Sci 127(Pt 3):686–699
Murthy M, Garza D, Scheller RH, Schwarz TL (2003) Mutations in the exocyst component Sec5 disrupt neuronal membrane traffic, but neurotransmitter release persists. Neuron 37(3):433–447
Letinic K, Sebastian R, Toomre D, Rakic P (2009) Exocyst is involved in polarized cell migration and cerebral cortical development. Proc Natl Acad Sci U S A 106(27):11342–11347
Rogers KK, Wilson PD, Snyder RW, Zhang X, Guo W, Burrow CR, Lipschutz JH (2004) The exocyst localizes to the primary cilium in MDCK cells. Biochem Biophysical Res Commun 319(1):138–143
Liu Q, Tan G, Levenkova N, Li T, Pugh EN Jr, Rux JJ, Speicher DW, Pierce EA (2007) The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics 6(8):1299–1317
Andersen NJ, Yeaman C (2010) Sec3-containing exocyst complex is required for desmosome assembly in mammalian epithelial cells. Mol Biol Cell 21(1):152–164
Xiong X, Xu Q, Huang Y, Singh RD, Anderson R, Leof E, Hu J, Ling K (2012) An association between type Iγ PI4P 5-kinase and Exo70 directs E-cadherin clustering and epithelial polarization. Mol Biol Cell 23(1):87–98
Liu J, Zuo X, Yue P, Guo W (2007) Phosphatidylinositol 4,5-bisphosphate mediates the targeting of the exocyst to the plasma membrane for exocytosis in mammalian cells. Mol Biol Cell 18(11):4483–4492
Thapa N, Sun Y, Schramp M, Choi S, Ling K, Anderson RA (2012) Phosphoinositide signaling regulates the exocyst complex and polarized integrin trafficking in directionally migrating cells. Dev Cell 22(1):116–130
Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, Olvera J, Bafna V, Zaki MS, Abdel-Salam GH, Mansour LA, Selim L, Abdel-Hadi S, Marzouki N, Ben-Omran T, Al-Saana NA, Sonmez FM, Celep F, Azam M, Hill KJ, Gleeson JG (2012) Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med 4(138):138ra78
Simsek-Kiper PO, Jacob P, Upadhyai P, Taşkıran ZE, Guleria VS, Karaosmanoglu B, Imren G, Gocmen R, Bhavani GS, Kausthubham N, Shah H, Utine GE, Boduroglu K, Girisha KM (2022) Biallelic loss-of-function variants in EXOC6B are associated with impaired primary ciliogenesis and cause spondylo-epi-metaphyseal dysplasia with joint laxity type 3. Hum Mutat 43(12):2116–2129
Zuo X, Guo W, Lipschutz JH (2009) The exocyst protein Sec10 is necessary for primary ciliogenesis and cystogenesis in vitro. Mol Biol Cell 20(10):2522–2529
Zuo X, Fogelgren B, Lipschutz JH (2011) The small GTPase Cdc42 is necessary for primary ciliogenesis in renal tubular epithelial cells. J Biol Chem 286(25):22469–22477
Choi SY, Chacon-Heszele MF, Huang L, McKenna S, Wilson FP, Zuo X, Lipschutz JH (2013) Cdc42 deficiency causes ciliary abnormalities and cystic kidneys. J Am Soc Nephrol 24(9):1435–1450
Feng S, Knödler A, Ren J, Zhang J, Zhang X, Hong Y, Huang S, Peränen J, Guo W (2012) A Rab8 guanine nucleotide exchange factor-effector interaction network regulates primary ciliogenesis. J Biol Chem 287(19):15602–15609
Lai YC, Kondapalli C, Lehneck R, Procter JB, Dill BD, Woodroof HI, Gourlay R, Peggie M, Macartney TJ, Corti O, Corvol JC, Campbell DG, Itzen A, Trost M, Muqit MM (2015) Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. EMBO J 34(22):2840–2861
Tanaka T, Goto K, Iino M (2017) Diverse functions and signal transduction of the exocyst complex in tumor cells. J Cell Physiol 232(5):939–957
Issaq SH, Lim KH, Counter CM (2010) Sec5 and Exo84 foster oncogenic ras-mediated tumorigenesis. Mol Cancer Res 8(2):223–231
Yan C, Theodorescu D (2018) RAL GTPases: biology and potential as therapeutic targets in cancer. Pharmacol Rev 70(1):1–11
Apken LH, Oeckinghaus A (2021) The RAL signaling network: cancer and beyond. Int Rev Cell Mol Biol 361:21–105
Sugihara K, Asano S, Tanaka K, Iwamatsu A, Okawa K, Ohta Y (2002) The exocyst complex binds the small GTPase RalA to mediate filopodia formation. Nat Cell Biol 4(1):73–78
Finger FP, Novick P (2000) Synthetic interactions of the post-Golgi sec mutations of Saccharomyces cerevisiae. Genetics 156(3):943–951
Cao F, Miao Y, Xu K, Liu P (2015) Lethal (2) giant larvae: an indispensable regulator of cell polarity and cancer development. Int J Biol Sci 11(4):380–389
Wong TA, Fairn GD, Poon PP, Shmulevitz M, McMaster CR, Singer RA, Johnston GC (2005) Membrane metabolism mediated by Sec14 family members influences Arf GTPase activating protein activity for transport from the trans-Golgi. Proc Natl Acad Sci USA 102(36):12777–12782
Manzer KM, Fromme JC (2023) The Arf-GAP Age2 localizes to the late-Golgi via a conserved amphipathic helix. Mol Biol Cell 34(12):ar119
Bender L, Lo HS, Lee H, Kokojan V, Peterson V, Bender A (1996) Associations among PH and SH3 domain-containing proteins and Rho-type GTPases in Yeast. J Cell Biol 133(4):879–894
Audhya A, Loewith R, Parsons AB, Gao L, Tabuchi M, Zhou H, Boone C, Hall MN, Emr SD (2004) Genome-wide lethality screen identifies new PI4,5P2 effectors that regulate the actin cytoskeleton. EMBO J 23(19):3747–3757
van den Bogaart G, Meyenberg K, Risselada HJ, Amin H, Willig KI, Hubrich BE, Dier M, Hell SW, Grubmüller H, Diederichsen U, Jahn R (2011) Membrane protein sequestering by ionic protein-lipid interactions. Nature 479(7374):552–555
Abramson J, Adler J, Dunger J et al (2024) Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630:493–500
Onat OE, Gulsuner S, Bilguvar K, Nazli Basak A, Topaloglu H, Tan M, Tan U, Gunel M, Ozcelik T (2013) Missense mutation in the ATPase, aminophospholipid transporter protein ATP8A2 is associated with cerebellar atrophy and quadrupedal locomotion. Eur J Hum Genet 21(3):281–285
Damásio J, Santos D, Morais S, Brás J, Guerreiro R, Sardoeira A, Cavaco S, Carrilho I, Barbot C, Barros J, Sequeiros J (2021) Congenital ataxia due to novel variant in ATP8A2. Clin Genet 100(1):79–83
Basante-Bedoya MA, Bogliolo S, Garcia-Rodas R, Zaragoza O, Arkowitz RA, Bassilana M (2022) Two distinct lipid transporters together regulate invasive filamentous growth in the human fungal pathogen Candida albicans. PLoS Genet 18(12):e1010549
Kim YG, Sohn EJ, Seo J, Lee KJ, Lee HS, Hwang I, Whiteway M, Sacher M, Oh BH (2005) Crystal structure of bet3 reveals a novel mechanism for Golgi localization of tethering factor TRAPP. Nat Struct Mol Biol 12(1):38–45
Saito K, Tautz L, Mustelin T (2007) The lipid-binding SEC14 domain. Biochim Biophys Acta. 1771(6):719–726
Wright ME, Peters U, Gunter MJ, Moore SC, Lawson KA, Yeager M, Weinstein SJ, Snyder K, Virtamo J, Albanes D (2009) Association of variants in two vitamin e transport genes with circulating vitamin e concentrations and prostate cancer risk. Can Res 69(4):1429–1438
Schott D, Ho J, Pruyne D, Bretscher A (1999) The COOH-terminal domain of Myo2p, a yeast myosin V, has a direct role in secretory vesicle targeting. J Cell Biol 147(4):791–808
Takagishi Y, Murata Y (2006) Myosin Va mutation in rats is an animal model for the human hereditary neurological disease, Griscelli syndrome type 1. Ann N Y Acad Sci 1086:66–80
Cağdaş D, Ozgür TT, Asal GT, Tezcan I, Metin A, Lambert N, Basile GS, Sanal O (2012) Griscelli syndrome types 1 and 3: analysis of four new cases and long-term evaluation of previously diagnosed patients. Eur J Pediatr 171(10):1527–1531
Puerner C, Serrano A, Wakade RS, Bassilana M, Arkowitz RA (2021) A myosin light chain is critical for fungal growth robustness in Candida albicans. mBio. 12(5):e0252821
Dong G, Hutagalung AH, Fu C, Novick P, Reinisch KM (2005) The structures of exocyst subunit Exo70p and the Exo84p C-terminal domains reveal a common motif. Nat Struct Mol Biol 12(12):1094–1100
Martin TD, Samuel JC, Routh ED, Der CJ, Yeh JJ (2011) Activation and involvement of Ral GTPases in colorectal cancer. Cancer Res 71(1):206–215
Sowalsky AG, Alt-Holland A, Shamis Y, Garlick JA, Feig LA (2011) RalA function in dermal fibroblasts is required for the progression of squamous cell carcinoma of the skin. Cancer Res 71(3):758–767
Caballero-Lima D, Sudbery PE (2014) In Candida albicans, phosphorylation of Exo84 by Cdk1-Hgc1 is necessary for efficient hyphal extension. Mol Biol Cell 25(7):1097–1110
Coulter ME, Musaev D, DeGennaro EM et al (2020) Regulation of human cerebral cortical development by EXOC7 and EXOC8, components of the exocyst complex, and roles in neural progenitor cell proliferation and survival. Genet Med 22(6):1040–1050
Ullah A, Krishin J, Haider N, Aurangzeb B, Abdullah SS, Ahmad W, Hansen T, Basit S (2022) A novel nonsense variant in EXOC8 underlies a neurodevelopmental disorder. Neurogenetics 23(3):203–212
Bassilana M, Hopkins J, Arkowitz RA (2005) Regulation of the Cdc42/Cdc24 GTPase module during Candida albicans hyphal growth. Eukaryot Cell 4(3):588–603
Wang Z, Oh E, Thurmond DC (2007) Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J Biol Chem 282(13):9536–9546
Audhya A, Emr SD (2002) Stt4 PI 4-kinase localizes to the plasma membrane and functions in the Pkc1-mediated MAP kinase cascade. Dev Cell 2(5):593–605
Qin H, Carr HS, Wu X, Muallem D, Tran NH, Frost JA (2005) Characterization of the biochemical and transforming properties of the neuroepithelial transforming protein 1. J Biol Chem 280(9):7603–7613
Gilcrease MZ, Kilpatrick SK, Woodward WA, Zhou X, Nicolas MM, Corley LJ, Fuller GN, Tucker SL, Diaz LK, Buchholz TA, Frost JA (2009) Coexpression of alpha6beta4 integrin and guanine nucleotide exchange factor Net1 identifies node-positive breast cancer patients at high risk for distant metastasis. Cancer Epidemiol Biomarkers Prev 18(1):80–86
Carvajal JJ, Pook MA, dos Santos M, Doudney K, Hillerman R, Minogue S, Williamson R, Hsuan JJ, Chamberlain S (1996) The Friedreich’s ataxia gene encodes a novel phosphatidylinositol-4- phosphate 5-kinase. Nat Genet 14(2):157–162
Migliorini G, Fiege B, Hosking FJ et al (2013) Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood 122(19):3298–3307
Hoopes RR Jr, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, Simckes A, Tasic V, Toenshoff B, Suchy SF, Nussbaum RL, Scheinman SJ (2007) Dent Disease with mutations in OCRL1. Am J Hum Genet 76(2):260–267
Cho HY, Lee BH, Choi HJ, Ha IS, Choi Y, Cheong HI (2008) Renal manifestations of Dent disease and Lowe syndrome. Pediatr Nephrol 23(2):243–249
Hichri H, Rendu J, Monnier N, Coutton C, Dorseuil O, Poussou RV, Baujat G, Blanchard A, Nobili F, Ranchin B, Remesy M, Salomon R, Satre V, Lunardi J (2011) From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum Mutat 32(4):379–388
Acknowledgements
We thank Chris Stefan (University College London) for valuable comments and suggestions about the manuscript, and Dheva Setiaputra (SFU Canada) for discussions about the molecular modeling.
Funding
This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada (https://www.nserc-crsng.gc.ca/) awarded to CTB.
Author information
Authors and Affiliations
Contributions
All authors contributed to the writing of this manuscript. The initial draft of the manuscript was compiled by CTB, and all authors were involved in editing every version thereafter.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Not applicable.
Consent for publication
All authors read and approved the final manuscript for publication.
Consent to participate
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Volpiana, M.W., Nenadic, A. & Beh, C.T. Regulation of yeast polarized exocytosis by phosphoinositide lipids. Cell. Mol. Life Sci. 81, 457 (2024). https://doi.org/10.1007/s00018-024-05483-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05483-x