
Abstract
Accumulating evidence has consolidated the interaction between viral infection and host alternative splicing. Serine-arginine (SR) proteins are a class of highly conserved splicing factors critical for the spliceosome maturation, alternative splicing and RNA metabolism. Serine-arginine protein kinases (SRPKs) are important kinases that specifically phosphorylate SR proteins to regulate their distribution and activities in the central pre-mRNA splicing and other cellular processes. In addition to the predominant SR proteins, other cytoplasmic proteins containing a serine-arginine repeat domain, including viral proteins, have been identified as substrates of SRPKs. Viral infection triggers a myriad of cellular events in the host and it is therefore not surprising that viruses explore SRPKs-mediated phosphorylation as an important regulatory node in virus–host interactions. In this review, we briefly summarize the regulation and biological function of SRPKs, highlighting their involvement in the infection process of several viruses, such as viral replication, transcription and capsid assembly. In addition, we review the structure–function relationships of currently available inhibitors of SRPKs and discuss their putative use as antivirals against well-characterized viruses or newly emerging viruses. We also highlight the viral proteins and cellular substrates targeted by SRPKs as potential antiviral therapeutic candidates.
Similar content being viewed by others

Introduction
A multitude of studies have consolidated the interaction between virus infection and host alternative splicing [1, 2]. Serine-arginine (SR) proteins are a class of highly conserved splicing factors with one or two RNA recognition motifs (RRMs) at the N-terminal and a serine-arginine dipeptide highly repetitive domain (RS domain) at the C-terminal (Fig. 1A) [3]. As RNA binding proteins, SR proteins are critical for the spliceosome maturation, splice site selection, alternative splicing and RNA metabolism, including mRNA export, translation, localization and nonsense-mediated RNA decay [4,5,6,7,8]. Notably, distribution and activities of SR proteins are mainly regulated by the reversible phosphorylation within the RS domain [9], and two protein kinase families, cdc2-like kinases (CLKs) [10] and serine-arginine protein kinases (SRPKs) [11, 12], are predominantly involved in the sequential phosphorylation modification process. SRPKs phosphorylate SR proteins in the cytoplasm to promote their nuclear import and storage in interchromatin speckles, whereas CLKs further phosphorylate SR proteins to stimulate their translocation and participation in RNA splicing (Fig. 1B). Under stress stimuli, SRPKs also translocate to the nucleus to regulate the hyperphosphorylation of SR proteins. Therefore, CLKs and SRPKs-mediated phosphorylation cooperate to precisely mobilize the splicing functions of SR proteins.

SRPKs phosphorylate SR proteins. A Structures of SRPKs and SR proteins. SRPKs contain two well-conserved catalytic kinase domains, which is separated by a non-conserved insert domain. SR splicing factors (SRSFs) consist of one or two RNA recognition motifs (RRMs) and a C-terminal RS domain, which is sequentially phosphorylated by SRPKs and CLKs for nuclear transport and distribution. B Cytoplasmic SRPKs interact with the cochaperones hsp40/Aha1 and the hsp90/hsp70 chaperon complex through the insert domain (green curve) and phosphorylate SR proteins to promote their nuclear translocation and nuclear speckles formation with the help of transportin-SR2. In the nucleus, CLKs further phosphorylate SR proteins to facilitate their participation in RNA splicing. Under different stresses, SRPKs dissociate from their chaperones and translocate to the nucleus to regulate the hyperphosphorylation of SR proteins and consequently alternative splicing. P phosphorylation
SRPKs belong to the serine-arginine protein kinase family that specifically phosphorylate SR splicing factors (SRSFs) and regulate the crucial pre-mRNA splicing process [11, 12]. To date, three members of SRPKs have been characterized, including the well-investigated SRPK1 (or its yeast analogue Sky1p) [13, 14], SRPK2 [15] and the less understood SRPK3 [16]. Apart from the prevailed canonical SRSFs (named SRSF1–12 according to their chronological order of discovery), other cytoplasmic proteins containing the RS domain have been identified as substrates of SRPKs, including viral proteins. Phosphorylation is an important regulatory mechanism leveraged by viruses to modulate the subcellular location, stability and activity of viral proteins and their interactions with host cellular proteins, which in turn facilitates viral infection and pathogenesis. It is therefore reasonable that viruses explore SRPKs-mediated phosphorylation of viral proteins or cellular substrates, such as SRSFs, to manipulate the alternative splicing of viral and host cell mRNA transcripts and to establish an appropriate microenvironment for viral replication and assembly. In this review, we highlight the complex contributions of SRPKs in viral infection and discuss the potential of SRPKs, viral proteins targeted by SRPKs, and SRSFs as therapeutic antiviral candidates.
Regulation and function of SRPKs
SRPKs consist of two conserved catalytic domains and a large non-conserved insert domain that separates the bipartite kinase domains [17]. The insert domain binds directly to the cochaperones Aha1 and Hsp40, mediating the dynamic interaction between SRPKs and the major heat-shock complex Hsp70/Hsp90 and anchoring SRPKs in the cytoplasm (Fig. 1B) [18,19,20]. Accordingly, cytoplasmically localized SRPKs predominantly phosphorylate newly synthesized SR proteins and other cytoplasmic RS-like domain-containing proteins (e.g., ZO-2, RNF12) [21, 22], and facilitate their nuclear import via transportin-SR2 [23, 24]. On the other hand, nuclear SRPKs collaborate with CLKs to tightly control the phosphorylation levels of SR proteins and modulate the efficient splicing pattern of several genes [25]. Under various external signals, such as osmotic stress [19], growth factors [20], cell cycle signals [18] and genotoxic agents [26], SRPKs are released from the Hsp70/Hsp90 chaperone complex and translocate to the nucleus to stimulate the expression of splicing isoforms conducive to cell growth via phosphorylation of SR proteins [20, 27], or to regulate lamin B receptor (LBR)-mediated chromatin reorganization [28, 29]. However, the unbalanced and aberrant increase in nuclear SRPKs is generally believed to cause the hyperphosphorylation of SR proteins and LBR, leading to splicing factor aggregation, splicing inhibition and chromatin segregation from the nuclear envelope [18, 29]. Notably, the transient interaction of nuclear matrix-localized SRPK1 with scaffold attachment factors (SAFB1/2) [30, 31], or the RNA binding protein TAF15 [32], in the nucleus impairs its enzymatic activity. When exposed to hypoxia, SRPK1 rapidly dissociates from SAFB1/2 and transfers to the cytoplasm, resulting in the dephosphorylation of SRSFs and the production of splicing variants.
In addition to these interacting proteins, post translational modifications, such as phosphorylation, acetylation and O-GlcNAcylation, have also been shown to regulate the kinase activity and nuclear translocation of SRPKs (Fig. 2). For example, casein kinase 2 (CK2) can phosphorylate SRPK1 at Ser51 to enhance its catalytic activity approximately sixfold in vitro [33]. Akt phosphorylates SRPK2 at Thr492 to promote its nuclear import and regulate cell cycle and cell death [34], whereas SRPK1 is phosphorylated at Thr326 and Ser587 in response to epidermal growth factor signaling [20]. Ribosomal S6 kinase 1 (S6K1) phosphorylates SRPK2 at Ser494 and cooperates with CK1-mediated phosphorylation at Ser497 to promote lipid biosynthesis [27]. In addition, the DNA damage response kinases ATM/ATR phosphorylate SRPK1 at Thr326 and Ser408 to adapt genotoxic stress [35]. Furthermore, the acetyltransferase Tip60 acetylates SRPK1 at Lys215/258/265/301/318/585/588 to promote its nuclear translocation and regulate cisplatin resistance [36, 37]. Finally, the O-GlcNAc transferase O-GlcNAcylates SRPK2 at Ser490, Thr492 and Thr498, triggering its binding to importin α and promoting the splicing of lipogenic pre-mRNAs [38]. Taken together, the cytoplasmic-nuclear shuttling of SRPKs mediated by post-transcriptional modifications represents a key approach for cells to respond to different stimuli.

Regulation of SRPKs. Stimuli-responsive kinases phosphorylate/acetylate/O-GlcNAcylate SRPKs at different amino acids, mainly within the insert domain, leading to the dissociation of SRPKs from the Hsp90/hsp70 complex and subsequent cytoplasm-nucleus translocation, which in turn modulate alternative splicing to achieve different outcomes. Transient interactions of SRPKs with nuclear SAFB1/2 and TAF15 further suppress SRPKs activity. Under stress, SRPKs can be released from these inhibitory interactions into the cytoplasm. P phosphorylation, Ac acetylation
Remarkably, SRPKs, initially considered to be phosphorylation factors of SR proteins during mitosis, are nowadays gradually being recognized as multifunctional proteins involved in a variety of cellular processes [12, 39]. Most SR proteins, including typical SRSFs and atypical SR splicing factors, have been confirmed to be substrates of SRPKs, allowing SRPKs to accomplish various mRNA splicing events. In addition, there are more than 100 RS or RS-like domain-containing proteins [40], suggesting that SRPKs may phosphorylate these potential targets to exert diverse cellular functions, such as mRNA maturation, spermiogenesis, cell cycle progression, chromatin reorganization and innate immunity [12, 41]. For example, SRPKs phosphorylate P1 protamine and LBR to regulate spermiogenesis [42]. SRPKs may modulate energy homeostasis and glucose/lipid metabolism through phosphorylating PPARgamma coactivator-1alpha (PGC-1α) [12]. Notably, there is growing amount of evidence that SRPKs are involved in various human diseases, including neurodegeneration [43], arthritis [44], atherosclerosis [45, 46], and cancer [47, 48]. A prominent example is that SRPKs-mediated alternative splicing facilitates the transition of vascular endothelial growth factor (VEGF) from anti-angiogenic to pro-angiogenic isoforms, resulting in neovascularization and angiogenesis [45, 46]. Additionally, SRPKs are highly expressed in numerous epithelial cancers and regulate several signaling pathways, such as NF-κB, TGF-β and PI3K/AKT pathways, to affect tumor cell growth and chemoresistance, and are considered as prognostic biomarkers and potential therapeutic targets [35, 49,50,51].
Viral manipulation of SRPKs-mediated phosphorylation
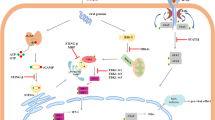
There is increasing evidence that, in addition to host cellular proteins, viral proteins containing RS or RS-like domains are also phosphorylated by SRPKs. Phosphorylation regulates various properties of viral proteins, such as activity, stability, subcellular location and interaction with protein partners or nucleic acids, and is essential for several critical steps of viral life cycle [52,53,54]. Substantial evidence suggests that different viruses interact with and hijack SRPKs to favor their replication, transcription and assembly (Table 1). Depending on the specific characteristics of the viral life cycle, SRPKs-mediated phosphorylation either induces the dissociation of viral proteins from the transcription complex/nucleic acids and their subsequent nuclear-cytoplasmic transport for transcription, or provides a suitable environment for viral capsid assembly and replication (Fig. 3). SRPKs also phosphorylate several SRSFs involved in the alternative splicing of viral and host cellular mRNAs.

Viral manipulation of SRPKs-mediated phosphorylation. A SRPKs-mediated phosphorylation regulates cytoplasmic-nuclear translocation of viral proteins, including HPV E2, VZV IE4, EBV BLRF2 and SARS-CoV N proteins. SRPKs also co-operate with viral proteins (HSV ICP27 and HPV1 E1^E4) to shut-off host splicing and promote viral transcription and replication. B SRPKs regulate VP30-mediated Ebola virus transcription. The transcription factor VP30 is dephosphorylated by PP2A and sequentially forms a complex with VP35 and polymerase L to initiate the primary transcription. VP30 is then phosphorylated by SRPK and dissociates from the RNA and polymerase complex to re-initiate the secondary transcription at the next transcription start site. C SRPKs regulate HBV capsid assembly. SRPKs bind to the CTD of the HBV core protein (HBc) to prevent its spontaneous assembly into RNA-filled capsids. The transient binding and appropriate phosphorylation by SRPKs allows the HBc-mediated adequate and organized HBV core formation, whereas hyperphosphorylation of HBc leads to the assembly of empty virions. Other viruses that interact with and modulate SRPKs function to favor their infection are not shown, including HCMV, HIV and HCV. CTD, C-terminal domain; P, phosphorylation; Me, methylation
Roles of SRPKs in dsDNA virus infection
Herpesviridae
Herpes simplex virus 1 (HSV-1) is a widely-spread double-stranded (ds) DNA virus and a member of the alpha-herpesvirus sub-family that infects and establishes latency in sensory neurons. The viral ICP27 protein, an mRNA export factor, plays a central role in regulating HSV-1 replication and triggering alternative splicing of host and viral transcripts [55], and its interaction with SRPK1 is one of the major mechanisms preventing host pre-mRNA maturation (Fig. 3A) [56]. ICP27 recruits SRPK1 to the nucleus, where it is directly phosphorylated by SRPK1. Concurrently, ICP27 inactivates SRPK1 and induces its nuclear accumulation, which in turn causes the hypophosphorylation of SRSFs (SRSF1 and SRSF5) and stalls host spliceosome assembly, favoring HSV-1 transcripts and viral replication [56]. Subsequently, ICP27 dissociates from the RNA and acts as a chaperone to promote the cytoplasmic export of viral RNA, a process that requiries arginine methylation. The arginine residue within the RGG motif (RS-like domain) is methylated by protein arginine methyltransferase 1 (PRMT1), the inhibition of which causes the hypomethylation of ICP27 and its earlier and more rapidly nuclear export, resulting in defective viral replication [57]. Notably, PRMT1-mediated methylation within the RGG motif also significantly reduces the SRPK1-ICP27 interaction and prevents the nuclear recruitment of SRPK1 [58, 59]. Mechanistically, the RGG motif exhibits a higher binding affinity to SRPK1 than to SR proteins, possibly explaining the function of ICP27 in preventing nuclear SRPK1 contact with hypophosphorylated SRSF1 and ensuring accurate splicing of viral genes with functional coding sequences [60, 61]. Therefore, the intricate interactions between ICP27, SRPK1 and PRMT1 occur sequentially in a specific order to ensure adequate viral replication and shut-off of host cellular splicing.
In addition to HSV-1 ICP27, SRPK1 has also been shown to interact with viral IE4 protein, an ICP27 homologue encoded by another alpha-herpesvirus, varicella-zoster virus (VZV) [62]. VZV IE4 protein is a chaperone that exports viral mRNA transcripts and interacts with SRSFs (SRSF1, SRSF3 and SRSF7) to act as a transcriptional activator. In the cytoplasm, SRPK1 binds to and phosphorylates IE4, triggering its dissociation from the exported mRNA and its cytoplasm-to-nucleus reshuttling (Fig. 3A). Furthermore, SRPK1 is involved in the infection of beta-herpesvirus human cytomegalovirus (HCMV) [63]. HCMV infection progressively triggers the accumulation of cytoplasmic SRPK1, which may allow efficient egress of viral nucleocapsids and continued RNA splicing during late HCMV infection. Interestingly, SRPK2 has been shown to interact with BLRF2, a tegument protein of the gamma-herpesvirus Epstein-Barr virus (EBV) [64]. BLRF2 either serves as a platform for tegument organization and capsid formation, or is essential for EBV replication by an undefined mechanism. SRPK2 phosphorylates BLRF2 within the RS motif (Ser148 and Ser150) to promote its nuclear localization, and depletion of this phosphorylation abrogates the critical and supportive function of BLRF2 in EBV replication. Finally, EBV EB2 protein, an ICP27 homologue belonging to the mRNA export factor, also interacts with SRSF1, SRSF3, and SRSF7 to stimulate cytoplasmic viral RNA accumulation and alternative splicing [65, 66]. Whether EB2 cooperates with SRPKs to modulate SRSF activity and viral transcription is unclear.
Papillomaviridae
Human papillomaviruses (HPVs) are non-enveloped dsDNA viruses that infect the oropharynx and anogenital tract to cause hyperproliferative warts and neoplasia. Although the pathogenesis of different HPV genotypes (Alpha, Beta, and Mu) is heterogeneous, the molecular mechanism of the viral life cycle is conservative, which is dependent on keratinocyte differentiation and requires the intervention of SRSFs-mediated HPV RNA splicing to balance the production of viral early (in undifferentiated cells) and late (in differentiated keratinocytes) proteins [67, 68]. Notably, phosphorylation of SRSFs by SRPK1 is usurped by some HPVs, and the interaction between SRPKs and HPV viral proteins is more complicated (Fig. 3A) [69]. During the virion-producing (late) phase of HPV, viral protein E4 is highly expressed to perform multiple roles, such as inducing cell cycle arrest and reorganizing the keratin network. E4 is derived from an E1^E4 fusion protein by proteolysis and sequesters various kinases, including SRPK1, to regulate late phase viral infection. It has been showed that HPV1 E1^E4 protein directly binds to and inhibits SRPK1 activity, reducing the phosphorylation of SRSFs (e.g., SRSF1, SRSF3, SRSF4 and SRSF7) and promoting the production of alternative viral RNA isoforms [70, 71]. Besides, SRPK1 facilitates the nuclear-to-cytoplasmic translocation of the HPV1 E2 protein, a viral DNA binding factor regulating viral replication and transcription partially through upregulating SRSFs. SRPK1 phosphorylates the RS domain within the hinge region of HPV1 E2, whereas E1^E4 inhibits such phosphorylation and retains E2 in the nucleolus to modulate post transcriptional expression of viral gene [70]. Likewise, SRPK1-mediated phosphorylation also transfers HPV5 E2 protein from the nucleus to the cytoplasm [23, 72], or binds to HPV8 E2 with unclear consequences [69]. In contrast, HPV16 E2 is able to enhance the expression of SRPK1 [73], which in turn phosphorylates and stabilizes the splicing factor SRSF1 to promote the production of viral late proteins and the completion of the HPV infection cycle in differentiated keratinocytes. Further works are required to prove whether E2 binds directly to the SRPK1 promoter or interacts indirectly with other transcription factors, such as WT1, to activate SRPK1 expression [74].
Roles of SRPKs in (+)ssRNA virus infection
Coronaviridae
The outbreak of the emerged severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused global pandemic with huge economic losses and human casualties. The nucleocapsid (N) protein of coronavirus (CoV) is a highly conserved, multiple functional and abundant RNA-binding protein responsible for genomic RNA encapsidation [75]. Besides, the N protein undergoes liquid–liquid phase separation to facilitate capsid assembly and to cluster the RNA polymerase complex for viral transcription [76, 77]. In addition, the N protein suppresses the cell-intrinsic antiviral immune defense mechanism RNAi and RIG-I-mediated innate immune response [78, 79]. Importantly, the above functions of N protein are affected by phosphorylation, particularly of its RS domain, which is located at the N-terminal domain (NTD) and determines its binding affinity to RNA and DNA [80]. During infection with the beta-coronavirus SARS-CoV-1, several kinases, such as CDK, GSK, CK2 and SRPK1, have been demonstrated to phosphorylate the N protein, at least in vitro [81]. SRPK1-mediated phosphorylation within the RS motif mobilizes N protein translocation to the cytoplasm, prevents its aggregation in stress granules and regulates its translational suppressive activity against host cells in the cytoplasm (Fig. 3A), whereas phosphorylation of the N protein by other kinases stimulates its cytoplasmic transport with unclear functional significance [82]. Besides, SRPK1-mediated extensive phosphorylation of newly synthesized N proteins prevents their aggregation and oligomerization in stress granules, leading to the soluble dimers favored for viral assembly [83]. In addition, SRPK1 and SRPK2 also phosphorylate the SARS-CoV-2 N protein at Ser188 and Ser206 to promote viral replication [84]. Through high-throughput screening, Alectinib, an FDA-approved inhibitor of anaplastic lymphoma kinase (ALK), is confirmed to inhibit SRPKs, resulting in decreased viral propagation and N protein phosphorylation [84]. Alectinib also dramatically inhibits the infection of alpha-coronavirus HCoV-229E, implying the extensively requirement of SRPK1/2 activity in both alpha- and beta-coronavirus life cycle. How insufficient phosphorylation of N protein compromises viral replication remains unclear, and one possibility is that N protein suppresses the host antiviral immune response [85]. Interestingly, non-COVID-19 patients show lower expression of SRPK2 when compared to COVID-19 patients, further suggesting the necessity of SRPKs for SARS-CoV-2 infection [86].
Flaviviridae
Hepatitis C virus (HCV) is an enveloped single-stranded RNA virus of the Flaviviridae family and is known for its pathogenic role in chronic liver diseases. In the case of HCV infection, alternative splicing has been demonstrated to shape the host antiviral immune response [87, 88]. Splicing factors generate different isoforms of interferon stimulated genes and human leukocyte antigen to significantly influence HCV infection and HCV-related carcinogenesis. On the other hand, HCV core protein, which is critical for nucleocapsid formation, viral genome encapsidation and interference with host cellular machinery, interacts with splicing factor DDX3 [89, 90], a DEAD-box RNA helicase involved in mRNA splicing and translation, to cooperatively activate viral polyprotein translation and RNA replication [91]. In addition to DDX3, SRPK1 and SRPK2 have also been shown to colocalize with HCV core protein and NS5A (an essential component of viral replication) in the perinuclear area, and inhibition of SRPKs by the chemical inhibitor SRPIN340 or by siRNA significantly reduces HCV replication [92]. Since phosphorylation of the core protein and NS5A regulates their subcellular localization and translational or replicative activity [93,94,95], it is unclear whether SRPKs directly phosphorylate core and NS5A protein to facilitate HCV infection. SRPKs may also phosphorylate other splicing factors (e.g., DDX3, DDX17) that contribute to HCV replication and pathogenesis of [96]. Furthermore, a recent work reported that HCV infection upregulates SRPK2 expression by downregulating the expression of a long non-coding RNA, Linc-Pint, which interacts with SRPK2 and suppresses its protein level. SRPK2 further promotes the phosphorylation of SRSFs (SRSF1, SRS4, SRSF5, and SRSF6) for the efficient splicing of several key genes involved in lipogenesis, such as ATP citrate lyase (ACLY) and fatty acid synthase (FASN), which in turn enhances de-novo lipogenesis to favor virus replication and liver disease [97]. Linc-Pint is regulated by p53 [98], and it is certainly conceivable that HCV core protein or NS5A may interfere with p53 signaling to inhibit Linc-Pint expression [99, 100].
Roles of SRPKs in (−)ssRNA virus infection
Filoviridae
Ebola virus is a filovirus that causes severe haemorrhagic fever, with a fatality rate of 34–81%. Once Ebola virus enters the cytoplasm via micropinocytosis, the release of the viral ribonucleoprotein (RNP) complex (consisting of the viral genome, transcription factor VP30, polymerase L, cofactor VP35, and nucleoprotein NP) promotes the production of all viral mRNAs (primary transcription) [101]. The newly produced viral mRNAs are then translated into various viral proteins, further facilitating the secondary transcription of viral mRNAs, genome replication, RNP complex formation as well as genomic RNA encapsidation in inclusion bodies. In particular, VP30 is indispensable for viral repetitive transcription, and reversible phosphorylation/dephosphorylation circuit of VP30 [102], mainly controlled by the cellular kinase phosphatase PP2A and SRPKs, modulates its subcellular location and binding affinity to the RNP complex [103,104,105]. VP30 is initially recruited and dephosphorylated by the cellular phosphatase PP2A interacting with NP, which in turn facilitates the replicative complex formation of VP30 with polymerase L and cofactor VP35 to initiate the primary transcription [106] (Fig. 3B). Upon completion, SRPK1 and SRPK2 phosphorylate VP30 at Ser29 within the ‘RXXS’ motif, and sequentially disperse it from the polymerase complex and RNA, allowing PP2A-mediated rapidly dephosphorylation of VP30 and ultimately redirection of VP30 to the secondary transcription start site to reinitiate the secondary transcription within inclusion bodies [105]. Therefore, SRPKs dynamically maintain the appropriate phosphorylation status of VP30 and ensure VP30-mediated effective transcription. Furthermore, VP30 is associated with the nucleocapsid [107], and it is uncertain whether SRPKs phosphorylate the nucleocapsid to promote capsid assembly.
Roles of SRPKs in reverse transcribing virus infection
Retroviridae (+ ssRNA-RT)
Human immunodeficiency virus (HIV) is a member of the Retroviridae family and its dynamic propagation and infectivity are dramatically affected by SR protein-mediated alternative splicing of viral RNAs [108]. Different forms of viral transcripts are produced to generate the nine HIV proteins: unspliced gRNA encodes Gag and Pol; fully spliced viral mRNA encodes Tat, Rev, and Nef; and the singly spliced mRNA (containing functional intron) generates Vif, Vpr, Vpu, and Env. In addition, more than 70 viral transcripts generated by suboptimal splicing have been identified to produce the viral proteins described above, in particularly Tat and Rev, two proteins critical for HIV replication. SR proteins regulate HIV gene expression at multiple steps, including transcription capping, splicing, 3′end formation, nuclear export and translation [109]. Consequently, modulation of SR protein expression and activity dramatically alters the extent of HIV RNA splicing and affects viral replication. For example, overexpression of the SR proteins SRSF1 (ASF/SF2), SRSF2 (SC35) or SRFS7 (9G8) suppresses HIV production, whereas overexpression of SRSF4 (SRp75), SRSF5 (SRp40) or SRSF6 (SRp55) exerts conversely functions that enhance HIV production and infectivity [110, 111]. Accordingly, HIV infection causes the dephosphorylation/abundance of most SR proteins, with the exception of SRSF4. HIV mobilizes SRPK2, and possibly SRPK1, to stabilize and phosphorylate SRSF4 to guarantee HIV-1 efficient replication [112]. In addition, depletion of SRPK1 has been shown to suppress HIV gene expression and the production of the viral structural proteins Gag and Env [113]. SRPK1 maintains HIV RNA levels and affects the transcriptional initiation of viral RNAs possibly by regulating the phosphorylation status of SRSF10 to modulate RNA splicing. Furthermore, a quantitative phosphorylation proteomics analysis indicates that the HIV accessory protein Vpr can phosphorylate and inhibit SRPK activity [114]. Future experiments are required to confirm the Vpr-SRPK interaction and to examine whether SRPK is involved in Vpr-mediated cell cycle regulation.
Hepadnaviridae (dsDNA-RT)
Hepatitis B virus (HBV), a small DNA virus of the Hepadnaviridae family, causes hepatitis and is causally associated with hepatocellular carcinoma. Unlike HIV, HBV does not require alternative splicing to produce viral proteins, although spliced viral transcripts exist [115]. HBV pregenomic RNA (pgRNA) is reverse transcribed into a circular and partially double-stranded DNA molecule after being packaged into the newly assembled capsid, a process predominantly regulated by the HBV core protein (HBc). Similar to the capsid proteins (N or core protein) of other viruses, HBc controls viral capsid assembly and persistent and effective HBV infection [116]. HBc activity is also regulated by phosphorylation, particularly phosphorylation within the RS domain located in the C terminal domain (CTD) of HBc [117, 118]. Notably, several host cellular kinases have been implicated in the phosphorylation of HBc, such as Cdc2, Cdk2, PKC and SRPKs, of which SRPKs are mainly responsible for the regulation of HBc phosphorylation and its function in vitro [118,119,120]. It is postulated that the encapsidation of HBV pgRNA requires an appropriate level of HBc phosphorylation, and SRPK1 and SRPK2 interact with and phosphorylate several serine residues within the CTD of HBc [120], acting as chaperones or neutralizers to ensure successful HBV genome packaging (Fig. 3C) [121,122,123]. Non-phosphorylated HBc has a higher binding affinity to RNA and readily self-assembles into RNA-filled capsids, which is inhibited by SRPKs-mediated phosphorylation [121]. In contrast, high phosphorylation of HBc by SRPKs leads to low RNA binding capacity and empty capsids/virions [122]. It is, therefore, speculated that after transient binding and phosphorylation of HBc, SRPKs dissociate from HBc to allow for slow HBc-mediated capsid formation and pgRNA incorporation [122, 123].
In addition to capsid assembly, the nucleic acid binding affinity of the CTD accredits HBc to participate in HBV DNA replication, and phosphorylation of different serine residues within the RS domain exhibits inconsistent but essential effects [124]. Interestingly, overexpression of both SRPK1 and SRPK2 has also been shown to suppress HBV replication by reducing pgRNA packaging without impact on core particle formation, and such suppressive effects of SRPKs do not depend on their phosphorylation activities on HBc [125]. Possible explanations are that SRPKs interfere with the interaction between HBc/pgRNA and HBc/viral DNA polymerase, or SRPKs are not the major kinases for HBc phosphorylation during HBV replication [126]. In addition, it remains to be determined whether SRPKs regulate the phosphorylation of splicing factor SRSFs to affect the binding of HBc to viral DNA/RNA and viral replication, as one recent work has shown that nuclear HBc interacts with several SR proteins and that inhibition of SRSF10, one of the substrates of SRPKs, regulates the amount of nascent HBV RNA [127]. Therefore, these results indicate the multiple and complex roles of SRPKs in the HBV life cycle. Finally, preliminary data from a recombinant co-expression system indicate that the core protein of the related duck hepatitis B virus (DHBV) is also highly phosphorylated by human SRPK1 [128]. Whether SRPK1 plays a key role in the virus-specific differential rates of nucleocapsid uncoating and pgRNA encapsidation remains to be determined.
SRPKs and SRPKs-associated viral or cellular proteins as emerging antiviral targets
It is clear that viruses hijack SRPKs-mediated phosphorylation of viral proteins and/or cellular substrates, mainly SRSFs, to manipulate multiple steps of the viral life cycle, such as alternative splicing, transcription, capsid assembly, viral pathogenesis, and even the host antiviral immune response. Therefore, SRPKs and SRPKs-targeted viral or cellular proteins are potent antiviral targets.
SRPKs as antiviral targets
Based on the essential role of SRPKs in pre-mRNA processing, inhibition of SRPKs has been suggested as a potential therapeutic opportunity against cancer and other diseases [11, 47, 48]. For example, EXN-107, a small molecule inhibitor of SRPK1 synthesized by the company Exonate (structure undisclosed), is currently in clinical trials (phase I/II, NCT04565756) to investigate its therapeutic efficacy against diabetic retinopathy. SCO-101, a potent inhibitor of SRPK1 and the drug efflux pump ABCG2 [129, 130], is currently in phase I/II clinical trials for the treatment of colorectal cancer (NCT04247256) and pancreatic ductal adenocarcinoma (NCT04652206).
To date, several small molecule inhibitors of SRPK (SRPKi), mainly derived from three scaffolds, have been developed, which bind irreversibly or reversibly to the ATP pocket. As presented in Fig. 4, the backbone of SRPKi, mainly the imine-nitrogen of benzimidazole [131] or the carbonyl group [132, 133], forms a crucial hydrogen (H)-bond with the backbone amide of Leu168 (SRPK1) in the hinge region, which contributes significantly to the activity of SRPKi and requires the flipping of the backbone carbonyl of Leu168. The presence of the hinge residue Gly169 further increases the flexibility and facilitates the flipping of the hinge region [134]. In addition, the interaction of residues Lys109 and Glu124 within the active site with the conserved Asp-Leu-Gly (DLG) motif at the adjacent ′allosteric′ site is critical for SRPK catalytic activity [132]. Accordingly, H-bond formation between Glu124 and the S1 substituent at one end of the backbone of SRPKi, as well as water molecule-mediated interaction between the S1 and Asp497/Leu498 of the DLG motif, significantly affects the inhibitory activity against SRPK1 [133]. At the opposite terminal end of SRPKi, key residues Tyr227, Ile228 and Leu231 of SRPK1 (Tyr239, Val240 and Met243 of SRPK2) within the insert helices adjacent to the ATP binding pocket influence both the activity and selectivity of SRPKi [132, 133]. The side chain of Tyr227 provides a steric constraint on the size of the S2 substitution. Alternatively, a covalent bond formation between Tyr227 and the S2 substituent (e.g., sulfonyl fluoride group) converts SRPKi into an irreversible inhibitor (e.g., SRPKIN-1) [133]. In addition, the larger side chains of Val240 and Met243 in SRPK2 create a smaller hydrophobic pocket and slightly change the binding orientation of SRPKi, resulting in reduced SRPK2 activity [132]. Interestingly, the S3 and S4 substituents may have opposite spatial positioning, with S3 interacting with the P-loop spatially in an upward direction and S4 partially interfacing with the DLG motif in a downward direction. Residues Ser92, Val94 and the P-loop backbone form a hydrophobic cavity oriented towards S3, and the improved contact between S3 and the P-loop, especially through aromatic stacking, significantly enhances inhibitor potency against SRPK1 [132]. Finally, a possible link between the S4 substituent and the DLG motif (Asp497/Leu498) or the lipophilic back pocket shaped by the side chains of Val145, Phe165, and Ala496 (SRPK1) is thought to increase the inhibitory activity of SRPKi on SRPK2 (to be determined).

Illustration of SRPK inhibitor binding in the ATP pocket. The crystal structure of SRPK1 in complex with inhibitor (PDB: 7ZKS) and key domains and relative residues around the substrate binding site are shown (SRPK1). Top and side views of the SRPK inhibitor in the ATP binding site are also presented. The grey parallelogram represents the backbone of the inhibitor, and the S1–S4 represent different substituents. Their interactive residues or domains of SRPK1 are shown
The structure–function relationships of SRPKi are summarized in Fig. 5, and their potent and efficient antiviral activities are shown in Table 2. The isonicotinamide compound SRPIN340 was firstly identified as an ATP-competitive inhibitor that covalently binds to the ATP binding pocket to inhibit SRPKs activities [112] (Fig. 5A). SRPIN340 exhibits high specificity for SRPK1/2 and does not significantly inhibit other SR protein-related kinases (e.g., CLK1 and CLK4) and Ser/Thr kinases. The half maximal inhibitory concentrations (IC50) of SRPIN340 against SRPK1 and SRPK2 are 0.89 μM and 7.4 μM, respectively [132]. SRPIN340 has also been very early shown to efficiently inhibit viral replication and production of HCV, Sindbis virus, CoVs, and Ebola virus (Table 2) [84, 92, 105, 112]. SPHINX and its derivatives SPHINX-1, SPHINX-2 and SPHINX-31, are other ATP-competitive inhibitors that share the same scaffold with SRPIN340 [132, 135]. Due to the interactions of different S3 substituent moieties with the P-loop of SRPK1, the SPHINX compounds exhibit higher activity and selectivity for SRPK1 over SRPK2 with a much lower IC50 value than that of SRPIN340 [135]. As a result, SPHINX-31 has a higher antiviral activity against SARS-CoV-2 than SRPIN340 (Table 2) [84]. SPHINX derivatives are also currently in preclinical studies for the treatment of leukemia, solid tumors and eye diseases [136,137,138]. However, the SPHINXs also exhibit possible effects on kinases of the CMGC family (CDK, MAPK, GSK, and CLK) [135].

Structure–function relationships of SRPK inhibitors. The chemical structures and corresponding SRPK-targeting activities of small molecule inhibitors and their derivatives, mainly derived from three scaffolds (A–C), are shown. The grey cycle represents the structural backbone and the orange dashed line represents the H-bond. IC50 half maximal inhibitory concentration. n.d. not determined
In addition, Alectinib, an FDA-approved primary anaplastic lymphoma kinase (ALK) inhibitor for the treatment of non-small-cell lung cancer, also displays potent inhibitory capacity to target SRPK1 (IC50 = 11 nM) [133] (Fig. 5B). Recently, Alectinib has been shown to inhibit the infection of coronavirus HCoV-229E and SARS-CoV-2 and reduce the SRPK1/2-mediated N protein phosphorylation [84], which may partly contribute to the favorable outcome of COVID-19 patients [139, 140]. Notably, replacing the 4-morpholinopiperidine moiety in Alectinib with a smaller pyrazole ring (compound JH-VII-139-1) results in a tenfold increase in SRPK1-targeting activity (IC50 = 1.1 nM). However, the inhibitory activity is reduced by ~ 1000-fold when substituted with a dimethyl amino-piperidine ring (compound JH-VII-206-2). Apart from SRPKs, Alectinib and JH-VII-139-1 also show broad interactions with numerous additional targets and inhibit the catalytic activity of other kinases, such as ALK and CHK2, to a similar extent [133]. To improve selectivity, the pyrazole ring at the S2 substitution in JH-VII-139–1 is further replaced by a 3-benzene-sulfonyl fluoride to generate a more specific SRPK-targeting analogue, SRPKIN-1. The sulfonyl fluoride group covalently interacts with Tyr227 of SRPK1 (or Tyr239 of SRPK2), making SRPKIN-1 an irreversible SRPK inhibitor with an IC50 of 35.6 nM for SRPK1 and an IC50 of 98 nM for SRPK2 [133]. Notably, SRPKIN-1 substantially reduces ALK activity (IC50 = 195 nM) and has little effect on CLK1 (IC50 > 10 μM) in a cell-based KiNativ profiling assay. In Ba/F3 cells transduced with EML4-ALK, a widely used model for testing ALK inhibitors, SRPKIN-1 shows 50-fold lower activity while JH-VII-206-2 and Alectinib show potent inhibitory effects, suggesting that the applicable concentrations of SRPKIN-1 at the cellular level have no influence on ALK. In addition, SRPKIN-1 is more than 50-fold more potent than SRPIN340 at phosphorylating SRSFs in a cellular context (200 nM vs. 10 μM) [133]. Future work is encouraged to investigate whether SRPKIN-1 has more potent and higher antiviral activity.
Recently, a series of compounds sharing a common benzimidazole-pyrimidine scaffold have been shown to be potent and reversible SRPKi (Fig. 5C), of which MSC-1186 (compound 5 m) is the most efficient and highly selective inhibitor with an IC50 of 3 nM for SRPK1 and IC50 of 80 nM for SRPK2 [131]. The benzimidazole-pyrimidine backbone acts as a hinge-binding moiety that interacts with the hinge region and flips it to form the essential H-bond with the Leu168 backbone amide (SRPK1). There is also a specific intramolecular H-bond exists between the protonated benzimidazole 3N-atom and one sulfonamide O-atom, which allows the S4 substitution to adopt a conformation capable of interacting with the lipophilic back pocket and the DLG motif. Disruption of such an intramolecular H-bond results in a twofold loss of SRPK1 inhibitory activity, while SRPK2 activity is enhanced (compound 5h vs 6). Notably, MSC-1186 shows good selectivity (rarely off-target) than other SRPK inhibitors SRPIN340, SPHINX-31, and SRPKIN-1 in in vitro kinase screening assay [131]. Surprisingly, MSC-1186 has very low to negligible activity as a single agent, but shows a robust additive effect in combination with CLK inhibitor in phosphorylating SRSFs in a cellular context. Future work is encouraged to investigate its antiviral activity and potential clinical application.
SRPKs-targeted viral proteins as antiviral targets
In addition to SRPKs, SRPKs-targeted viral proteins also represent attractive therapeutic targets for the control of viral infections and virus-related diseases. SRPKs-mediated phosphorylation of the RS motif within viral capsid proteins (the NTD of CoV N protein and EBV BLRF2, or the CTD of HBV core protein), significantly affects their binding affinity to RNA/DNA, which is pivotal for viral RNP formation, genome encapsidation and replication. Accordingly, blocking the nucleic acid binding ability of viral capsid proteins may be a potent antiviral strategy [141]. Indeed, several compounds, such as N-(6-oxo-5,6-dihydrophenanthridin-2-yl)(N,N-dimethylamino) acetamide hydrochloride (PJ34) and 6-chloro-7-(2-morpholin-4-ylethylamino) quinoxaline-5,8-dione (H3), have been shown to target the RNA binding site within the NTD of the N protein to inhibit CoV replication [142, 143]. Alternatively, the compound 5-benzyloxygramine induces abnormal dimerization and aggregation of N protein to suppress RNP formation and MERS-CoV infection [144]. In addition, inhibition of the oligomerization of N protein by competing peptides is also presumed to be an effective approach in the fight against HIV infection [145]. Similarly, core proteins of HBV and HCV are involved in multiple steps of the viral cycle through their dimerization and interaction with other viral or cellular proteins, and their specific inhibitors have been developed as novel antiviral agents. For example, an NTD-derived peptide inhibitor dramatically inhibits the dimerization of HCV core protein and blocks HCV infectious production [146]. In the context of HBV, different chemotypes of core protein allosteric modulators (CpAMs) have been discovered to inhibit HBV replication and to treat chronic hepatitis B in clinical trials (reviewed in ref. [147]). All the CpAMs bind to the hydrophobic pocket between the interface of core protein dimers, and type I CpAMs accelerate core protein assembly into degradative polymers or aberrant capsids, whereas type II CpAMs promote the formation of empty capsids without pgRNA. CpAMs also disrupt the de novo cccDNA synthesis and DNA replication with unclear mode of action. Notably, several CpAMs, such as RO9389 (type I) [148] and JNJ-6379 (type II) [149], are under clinical investigation for chronic HBV infection and have shown beneficial effects in patients. It will be interesting to investigate whether these above described inhibitors regulate SRPKs to exert their antiviral effects.
In addition, SRPKs-mediated phosphorylation of the Ebola virus transcription factor VP30 from is essential for repetitive viral transcription [150], and compounds targeting VP30 phosphorylation status, such as okadaic acid [151], have been shown to impair Ebola virus infection. In addition, VP30 interacts with NP protein to modulate viral RNA synthesis and disruption of the VP30-NP interaction is also a potential antiviral treatment, as illustrated by the antiviral activity of an NP-competitive peptide mimic [152].
Furthermore, SRPKs-mediated phosphorylation of herpesviral mRNA export proteins (HSV ICP27 and VSV IE4) or HPV E2 protein regulates their nucleus-to-cytoplasm reshuttling and viral gene expression, which may also be considered as novel therapeutic candidates [153, 154]. One example is that a peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO) has been shown to inhibit HSV-1 replication by targeting ICP27 [155]. Further structure-based high-throughput screening and experimental validation may identify more potential inhibitors of these viral proteins.
Substrates of SRPKs as antiviral targets
As discussed above, SRSFs are mainly substrates of SRPKs and their appropriate phosphorylation levels are critical for viral infection, suggesting them as alternative candidates for antiviral therapy. For example, SRSF10 interacts with HBV core protein and the small molecule compound 1C8, which specifically inhibits SRSF10 phosphorylation, significantly reduces HBV nascent RNA production [127]. SRSF10 also affects HIV pre-mRNA splicing and 1C8 treatment reduces HIV replication [156, 157]. Cardiotonic steroids, such as digoxin and digitoxin, deplete the expression of SRSF3 and SRSF10 to alter viral alternative splicing and suppress HIV replication [158, 159]. In addition, SRSF5 directly enhances the pre-mRNA splicing of the M protein to promote influenza virus replication, which is further suppressed by anidulafungin, an FDA-approved antifungal drug that has been identified as an inhibitor of SRSF5 [160]. Anidulafungin also exhibits a broad-spectrum antiviral activity that effectively blocks the infection of Zika virus, HSV-1, SARS-CoV-2 and other viruses [161, 162], suggesting the functional involvement of SRSF5 in different viruses. Recently, several new inhibitors of SRSFs have gradually emerged, making it possible to target specific SRSFs in viral infections. For example, through high-throughput screening and modification, SFI003 is confirmed to be a specific inhibitor of SRSF3 that binds to SRSF3 and induces its neddylation-dependent degradation [163]. Indacaterol, an approved β2-adrenergic receptor agonist, is identified as an inhibitor of SRSF6 [164], and has been implicated in disrupting the interaction between ACE2 and the S protein of SARS-CoV-2 by virtual screening [165]. Future investigations into the understanding of the biological functions of SRSFs and SRPKs in the viral life cycle, and the development of novel specific inhibitors of SRSFs (e.g., SRSF2, SRSF4, SRSF7-9) may pave the way for clinical application of SRSFs inhibitors. Alternatively, antisense oligonucleotide-mediated downregulation of SRSFs is another exciting therapeutic avenue to be further explored.
Perspectives
Nowadays, the emergence of a variety of highly infectious and pathogenic viruses poses a serious threat to human health. How to find the effective antiviral drugs quickly and accurately is a major challenge, making drug repurposing an optimal alternative strategy. SRPKs are one of the key regulators of mRNA maturation and splicing, and their role in viral infection is receiving increasing attention. In this review, we briefly summarize the regulation and biological function of SRPKs, highlight their involvement in the infection process of several viruses and discuss the putative use of inhibitors targeting SRPKs and SRPKs-associated viral proteins or cellular substrates as potential antiviral strategies.
Although the cellular biological functions of SRPKs in RNA splicing have been recognized for many years, several questions remain unanswered. For example, whether other post-transcriptional modifications or interacting proteins regulate the activity and cellular location of SRPKs. Besides, SRPKs-mediated phosphorylation represents as a prevalent regulatory mechanism to influence the cellular location and transcriptional activity of RS domain-containing viral proteins. Different functional types of viral proteins, including capsid protein (e.g., CoV N protein, EBV BLRF2, HBV core protein), mRNA export factor (e.g., HSV ICP27, VSV IE4, HPV E2), and transcription factor (e.g., Ebola virus VP30), are phosphorylated and regulated by SRPKs. More interactive connections between viral proteins (e.g., capsids and transcriptional activators of other undisclosed viruses) and SRPKs may be found by analyzing phosphorylation proteomics. It is also unclear whether SRPKs influence viral infection by modulating the antiviral immune response [41, 166]. In addition to the above-mentioned viruses that have been clearly shown to interact with SRPKs, other viruses that usurp the cellular machinery mediated by SRSFs for their replication are emerging as being able to control SRPKs-mediated phosphorylation. For example, several SRSFs (e.g., SRSF2, SRSF3, and SRSF5) are essential for viral replication of influenza virus, a small RNA virus hijacking cellular splicing components to produce its M and NS proteins [160, 167,168,169]. It is, therefore, of interest to investigate whether influenza virus modulates the activity of SRPKs to maintain appropriate phosphorylation levels of SRSFs and to explore the possible antiviral activities of SRPK inhibitors. In addition, considering that CLKs and SRPKs jointly regulate the phosphorylation of SR proteins in the nucleus and a growing body of evidence shows the interplay between different viruses and CLKs [113, 169,170,171,172], inhibition of CLKs by inhibitors, or synergistically in combination with SRPKs inhibition, is novel robust therapeutic option for viral infections.
Furthermore, SRPKs and SRPKs-targeted viral proteins or cellular proteins (e.g., SRSFs) are potential antiviral targets. The feasibility of developing SRPKs inhibitors that specifically target unique virus–host interactions, as SRPKs interact directly with different viral proteins, needs to be validated. Targeting SRSFs with inhibitors, competing peptides, or antisense-oligonucleotides may also be an alternative and interesting strategy to counteract viral infection. Given the abnormally high expression of SRPKs and SRSFs in some cancers [49, 173], inhibitors of SRPKs and SRSFs may have both anticancer and antiviral effects in the case of oncogenic viruses (e.g., HPV, HBV). Future work is also warranted to verify the potential of SRPKs and SRSFs inhibitors in relevant anti-infective clinical applications. However, the existing SRPKs inhibitors developed in cancer therapy are both insufficiently potent and selective, with unclear selectivity towards other kinases. Although the inhibitor MSC-1186 exhibits high selectivity and activity towards SRPKs, its cellular potency and efficacy are limited [131]. Therefore, the development of more efficient and specific SRPKs inhibitors will enable more reliable investigation of the undiscovered biological functions of SRPKs and provide more options for antiviral treatment.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
References
Sehrawat S, Garcia-Blanco MA (2023) RNA virus infections and their effect on host alternative splicing. Antiviral Res 210:105503
Mann JT, Riley BA, Baker SF (2023) All differential on the splicing front: Host alternative splicing alters the landscape of virus-host conflict. Semin Cell Dev Biol S1084–9521(23):00021–00026
Shepard PJ, Hertel KJ (2009) The SR protein family. Genome Biol 10:242
Jeong S (2017) SR proteins: binders, regulators, and connectors of RNA. Mol Cells 40:1–9
Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP et al (2007) SR proteins function in coupling RNAP II transcription to Pre-mRNA splicing. Mol Cell 26:867–881
Müller-McNicoll M, Botti V, de Jesus Domingues AM, Brandl H, Schwich OD, Steiner MC et al (2016) SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev 30:553–566
Cáceres JF, Screaton GR, Krainer AR (1998) A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev 12:55–66
Zhang Z, Krainer AR (2004) Involvement of SR Proteins in mRNA Surveillance. Mol Cell 16:597–607
Zhou Z, Fu XD (2013) Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 122(3):191–207
Martín Moyano P, Němec V, Paruch K (2020) Cdc-like kinases (CLKs): biology, chemical probes, and therapeutic potential. Int J Mol Sci 21(20):7549
Tang J, Xie Y, Huang J, Zhang L, Jiang W, Li Z et al (2022) A critical update on the strategies towards small molecule inhibitors targeting Serine/arginine-rich (SR) proteins and Serine/arginine-rich proteins related kinases in alternative splicing. Bioorg Med Chem 70:116921
Giannakouros T, Nikolakaki E, Mylonis I, Georgatsou E (2011) Serine-arginine protein kinases: a small protein kinase family with a large cellular presence. FEBS J 278(4):570–586
Gui JF, Lane WS, Fu XD (1994) A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature 369(6482):678–682
Kuroyanagi N, Onogi H, Wakabayashi T, Hagiwara M (1998) Novel SR-protein-specific kinase, SRPK2, disassembles nuclear speckles. Biochem Biophys Res Commun 242(2):357–364
Wang HY, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC et al (1998) SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J Cell Biol 140:737–750
Xu Y, Yu W, Xiong Y, Xie H, Ren Z, Xu D et al (2011) Molecular characterization and expression patterns of serine/arginine-rich specific kinase 3 (SRPK3) in porcine skeletal muscle. Mol Biol Rep 38(5):2903–2909
Nikolakaki E, Sigala I, Giannakouros T (2022) Good cop, bad cop: The different roles of SRPKs. Front Genet 13:902718
Ding JH, Zhong XY, Hagopian JC, Cruz MM, Ghosh G, Feramisco J et al (2006) Regulated cellular partitioning of SR protein-specific kinases in mammalian cells. Mol Biol Cell 17(2):876–885
Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD (2009) Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev 23(4):482–495
Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H et al (2012) The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell 47:422–433
Quiros M, Alarcón L, Ponce A, Giannakouros T, González-Mariscal L (2013) The intracellular fate of zonula occludens 2 is regulated by the phosphorylation of SR repeats and the phosphorylation/O-GlcNAcylation of S257. Mol Biol Cell 24:2528–2543
Bustos F, Segarra-Fas A, Nardocci G, Cassidy A, Antico O, Davidson L et al (2020) Functional diversification of SRSF Protein Kinase to control ubiquitin-dependent neurodevelopmental signaling. Dev Cell 55:629-647.e7
Lai MC, Lin RI, Huang SY, Tsai CW, Tarn WY (2000) A human importin-beta family protein, transportin-SR2, interacts with the phosphorylated RS domain of SR proteins. J Biol Chem 275:7950–7957
Lai MC, Lin RI, Tarn WY (2001) Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc Natl Acad Sci USA 98:10154–10159
Aubol BE, Wu G, Keshwani MM, Movassat M, Fattet L, Hertel KJ et al (2016) Release of SR proteins from CLK1 by SRPK1: A symbiotic kinase system for phosphorylation control of Pre-mRNA splicing. Mol Cell 63:218–228
Sigala I, Koutroumani M, Koukiali A, Giannakouros T, Nikolakaki E (2021) Nuclear translocation of SRPKs is associated with 5-FU and cisplatin sensitivity in HeLa and T24 cells. Cells 10:759
Lee G, Zheng Y, Cho S, Jang C, England C, Dempsey JM et al (2017) Post-transcriptional regulation of De Novo lipogenesis by mTORC1-S6k1-SRPK2 signaling. Cell 171:1545–1558
Takano M, Koyama Y, Ito H, Hoshino S, Onogi H, Hagiwara M et al (2004) Regulation of binding of lamin B receptor to chromatin by SR protein kinase and Cdc2 kinase in Xenopus egg extracts. J Biol Chem 279:13265–13271
Sellis D, Drosou V, Vlachakis D, Voukkalis N, Giannakouros T, Vlassi M (2012) Phosphorylation of the arginine/serine repeats of lamin B receptor by SRPK1-insights from molecular dynamics simulations. Biochim Biophys Acta (BBA) General Subj 1820:44–55
Tsianou D, Nikolakaki E, Tzitzira A, Bonanou S, Giannakouros T, Georgatsou E (2009) The enzymatic activity of SR protein kinases 1 and 1a is negatively affected by interaction with scaffold attachment factors B1 and 2. FEBS J 276(18):5212–5227
Taze C, Drakouli S, Samiotaki M, Panayotou G, Simos G, Georgatsou E et al (2022) Short-term hypoxia triggers ROS and SAFB mediated nuclear matrix and mRNA splicing remodeling. Redox Biol 58:102545
Koukiali A, Daniilidou M, Mylonis I, Giannakouros T, Nikolakaki E (2022) SR protein kinase 1 inhibition by TAF15. Cells 12(1):126
Mylonis I, Giannakouros T (2003) Protein kinase CK2 phosphorylates and activates the SR protein-specific kinase 1. Biochem Biophys Res Commun 301:650–656
Jang SW, Liu X, Fu H, Rees H, Yepes M, Levey A et al (2009) Interaction of Akt-phosphorylated SRPK2 with 14-3-3 mediates cell cycle and cell death in neurons. J Biol Chem 284:24512–24525
Sigala I, Koutroumani M, Koukiali A, Giannakouros T, Nikolakaki E (2021) Nuclear translocation of SRPKs is associated with 5-FU and cisplatin sensitivity in HeLa and T24 cells. Cells 10(4):759
Wang C, Zhou Z, Subhramanyam CS, Cao Q, Heng ZSL, Liu W et al (2020) SRPK1 acetylation modulates alternative splicing to regulate cisplatin resistance in breast cancer cells. Commun Biol 3(1):268
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC et al (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840
Tan W, Jiang P, Zhang W, Hu Z, Lin S, Chen L et al (2021) Posttranscriptional regulation of de novo lipogenesis by glucose-induced O-Glcnacylation. Mol Cell 81:1890-1904.e7
Ghosh G, Adams JA (2011) Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J 278(4):587–597
Calarco JA, Superina S, O’Hanlon D, Gabut M, Raj B, Pan Q et al (2009) Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell 138(5):898–910
Nousiainen L, Sillanpää M, Jiang M, Thompson J, Taipale J et al (2013) Human kinome analysis reveals novel kinases contributing to virus infection and retinoic-acid inducible gene I-induced type I and type III IFN gene expression. Innate Immun 19:516–530
Papoutsopoulou S, Nikolakaki E, Chalepakis G, Kruft V, Chevaillier P, Giannakouros T (1999) SR protein-specific kinase 1 is highly expressed in testis and phosphorylates protamine 1. Nucleic Acids Res 27(14):2972–2980
Chan CB, Ye K (2013) Serine-arginine protein kinases: new players in neurodegenerative diseases? Rev Neurosci 24(4):401–413
Oltean S, Gammons M, Hulse R, Hamdollah-Zadeh M, Mavrou A, Donaldson L et al (2012) SRPK1 inhibition in vivo: modulation of VEGF splicing and potential treatment for multiple diseases. Biochem Soc Trans 40(4):831–835
Zhao N, Zhang J (2018) Role of alternative splicing of VEGF-A in the development of atherosclerosis. Aging (Albany NY) 10(10):2695–2708
Nowak DG, Amin EM, Rennel ES, Hoareau-Aveilla C, Gammons M, Damodoran G et al (2010) Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J Biol Chem 285:5532–5540
Czubaty A, Piekiełko-Witkowska A (2017) Protein kinases that phosphorylate splicing factors: roles in cancer development, progression and possible therapeutic options. Int J Biochem Cell Biol 91(Pt B):102–115
Corkery DP, Holly AC, Lahsaee S, Dellaire G (2015) Connecting the speckles: splicing kinases and their role in tumorigenesis and treatment response. Nucleus 6(4):279–288
Duggan WP, O’Connell E, Prehn JHM, Burke JP (2022) Serine-arginine protein kinase 1 (SRPK1): a systematic review of its multimodal role in oncogenesis. Mol Cell Biochem 477(10):2451–2467
van Roosmalen W, Le Dévédec SE, Golani O, Smid M, Pulyakhina I, Timmermans AM et al (2015) Tumor cell migration screen identifies SRPK1 as breast cancer metastasis determinant. J Clin Investig 125(4):1648–1664
Tzelepis K, De Braekeleer E, Aspris D, Barbieri I, Vijayabaskar MS, Liu WH et al (2018) SRPK1 maintains acute myeloid leukemia through effects on isoform usage of epigenetic regulators including BRD4. Nat Commun 9(1):5378
Jakubiec A, Jupin I (2007) Regulation of positive-strand RNA virus replication: the emerging role of phosphorylation. Virus Res 129:73–79
Keck F, Ataey P, Amaya M, Bailey C, Narayanan A (2015) Phosphorylation of single stranded RNA virus proteins and potential for novel therapeutic strategies. Viruses 7(10):5257–5273
Lee JY, Lucas WJ (2001) Phosphorylation of viral movement proteins–regulation of cell-to-cell trafficking. Trends Microbiol 9(1):5–8
Sandri-Goldin RM (2011) The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol 6:1261–1277
Sciabica KS, Dai QJ, Sandri-Goldin RM (2003) ICP27 interacts with SRPK1 to mediate HSV splicing inhibition by altering SR protein phosphorylation. EMBO J 22:1608–1619
Souki SK, Gerson PD, Sandri-Goldin RM (2009) Arginine methylation of the ICP27 RGG box regulates ICP27 export and is required for efficient herpes simplex virus 1 replication. J Virol 83:5309–5320
Souki SK, Sandri-Goldin RM (2009) Arginine methylation of the ICP27 RGG box regulates the functional interactions of ICP27 with SRPK1 and Aly/REF during herpes simplex virus 1 infection. J Virol 83:8970–8975
Souki SK, Hernandez FP, Sandri-Goldin RM (2011) Arginine methylation of the RGG box does not appear to regulate ICP27 import during herpes simplex virus infection. J Virol 85(13):6809–6813
Tunnicliffe RB, Hu WK, Wu MY, Levy C, Mould AP, McKenzie EA et al (2019) Molecular mechanism of SR protein kinase 1 inhibition by the herpes virus protein ICP27. MBio 10(5):e02551-e2619
Tang S, Patel A, Krause PR (2019) Hidden regulation of herpes simplex virus 1 pre-mRNA splicing and polyadenylation by virally encoded immediate early gene ICP27. PLoS Pathog 15:e1007884
Ote I, Lebrun M, Vandevenne P, Bontems S, Medina-Palazon C, Manet E et al (2009) Varicella-zoster virus IE4 protein interacts with SR proteins and exports mRNAs through the TAP/NXF1 pathway. PLoS ONE 4(11):e7882
Gaddy CE, Wong DS, Markowitz-Shulman A, Colberg-Poley AM (2010) Regulation of the subcellular distribution of key cellular RNA-processing factors during permissive human cytomegalovirus infection. J Gen Virol 91(Pt 6):1547–1559
Duarte M, Wang L, Calderwood MA, Adelmant G, Ohashi M, Roecklein-Canfield J et al (2013) An RS motif within the Epstein-Barr virus BLRF2 tegument protein is phosphorylated by SRPK2 and is important for viral replication. PLoS ONE 8(1):e53512
Juillard F, Bazot Q, Mure F, Tafforeau L, Macri C, Rabourdin-Combe C et al (2012) Epstein-Barr virus protein EB2 stimulates cytoplasmic mRNA accumulation by counteracting the deleterious effects of SRp20 on viral mRNAs. Nucleic Acids Res 40:6834–6849
Verma D, Bais S, Gaillard M, Swaminathan S (2010) Epstein-Barr virus SM protein utilizes cellular splicing factor SRp20 to mediate alternative splicing. J Virol 84:11781–11789
Graham SV (2017) The human papillomavirus replication cycle, and its links to cancer progression: a comprehensive review. Clin Sci 131:2201–2221
Graham SV, Faizo AAA (2017) Control of human papillomavirus gene expression by alternative splicing. Virus Res 231:83–95
Jang MK, Anderson DE, van Doorslaer K, McBride AA (2015) A proteomic approach to discover and compare interacting partners of papillomavirus E2 proteins from diverse phylogenetic groups. Proteomics 15:2038–2050
Prescott EL, Brimacombe CL, Hartley M, Bell I, Graham S et al (2014) Human papillomavirus type 1 E1^E4 protein is a potent inhibitor of the serine-arginine (SR) protein kinase SRPK1 and inhibits phosphorylation of host SR proteins and of the viral transcription and replication regulator E2. J Virol 88:12599–12611
Bell I, Martin A, Roberts S (2007) The E1^E4 protein of human papillomavirus interacts with the serine-arginine-specific protein kinase SRPK1. J Virol 81:5437–5448
Wang WS, Lee MS, Tseng CE, Liao IH, Huang SP, Lin RI et al (2009) Interaction between human papillomavirus type 5 E2 and polo-like kinase 1. J Med Virol 81:536–544
Mole S, Faizo AAA, Hernandez-Lopez H, Griffiths M, Stevenson A, Roberts S et al (2020) Human papillomavirus type 16 infection activates the host serine arginine protein kinase 1 (SRPK1)—splicing factor axis. J Gen Virol 101(5):523–532
Amin EM, Oltean S, Hua J, Gammons MVR, Hamdollah-Zadeh M et al (2011) Wt1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 20:768–780
Chang CK, Hsu YL, Chang YH, Chao FA, Wu MC, Huang YS et al (2009) Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: Implications for ribonucleocapsid protein packaging. J Virol 83:2255–2264
Carlson CR, Asfaha JB, Ghent CM, Howard CJ, Hartooni N, Safari M et al (2020) Phosphoregulation of phase separation by the SARS-CoV-2 N protein suggests a biophysical basis for its dual functions. Mol Cell 80:1092-1103.e1094
Savastano A, Ibáñez de Opakua A, Rankovic M, Zweckstetter M (2020) Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat Commun 11:6041
Cui L, Wang H, Ji Y, Yang J, Xu S, Huang X et al (2015) The Nucleocapsid protein of coronaviruses acts as a viral suppressor of RNA silencing in mammalian cells. J Virol 89:9029–9043
Zheng Y, Zhuang MW, Han L, Zhang J, Nan ML, Zhan P et al (2020) Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct Target Ther 5:299
Surjit M, Kumar R, Mishra RN, Reddy MK, Chow VT, Lal SK (2005) The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14-3-3-mediated translocation. J Virol 79:11476–11486
Wu CH, Yeh SH, Tsay YG, Shieh YH, Kao CL, Chen YS et al (2009) Glycogen synthase kinase-3 regulates the phosphorylation of severe acute respiratory syndrome coronavirus nucleocapsid protein and viral replication. J Biol Chem 284(8):5229–5239
Peng TY, Lee KR, Tarn WY (2008) Phosphorylation of the arginine/serine dipeptide-rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J 275:4152–4163
Nikolakaki E, Giannakouros T (2020) SR/RS motifs as critical determinants of coronavirus life cycle. Front Mol Biosci 7:219
Yaron TM, Heaton BE, Levy TM, Johnson JL, Jordan TX, Cohen BM et al (2022) Host protein kinases required for SARS-CoV-2 nucleocapsid phosphorylation and viral replication. Sci Signal 15(757):eabm0808
Oh SJ, Shin OS (2021) SARS-CoV-2 nucleocapsid protein targets RIG-I-like receptor pathways to inhibit the induction of interferon response. Cells 10:530
Molinero M, Gómez S, Benítez ID, Vengoechea JJ, González J, Polanco D et al (2022) Multiplex protein profiling of bronchial aspirates reveals disease-, mortality- and respiratory sequelae-associated signatures in critically ill patients with ARDS secondary to SARS-CoV-2 infection. Front Immunol 13:942443
Lin W, Zhu C, Hong J, Zhao L, Jilg N, Fusco DN et al (2015) The spliceosome factor SART1 exerts its anti-HCV action through mRNA splicing. J Hepatol 62(5):1024–1032
Tremblay MP, Armero VES, Allaire A, Boudreault S, Martenon-Brodeur C, Durand M et al (2016) Global profiling of alternative RNA splicing events provides insights into molecular differences between various types of hepatocellular carcinoma. BMC Genom 17:683
Mamiya N, Worman HJ (1999) Hepatitis C virus core protein binds to a DEAD box RNA helicase. J Biol Chem 274:15751–15756
Owsianka AM, Patel AH (1999) Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology 257:330–340
Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T et al (2007) DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J Virol 81:13922–13926
Karakama Y, Sakamoto N, Itsui Y, Nakagawa M, Tasaka-Fujita M, Nishimura-Sakurai Y et al (2010) Inhibition of hepatitis C virus replication by a specific inhibitor of serine-arginine-rich protein kinase. Antimicrob Agents Chemother 54(8):3179–3186
Shih CM, Chen CM, Chen SY, Lee YH (1995) Modulation of the trans-suppression activity of hepatitis C virus core protein by phosphorylation. J Virol 69:1160–1171
Huang Y, Staschke K, De Francesco R, Tan SL (2007) Phosphorylation of hepatitis C virus NS5A nonstructural protein: a new paradigm for phosphorylation-dependent viral RNA replication? Virology 364:1–9
Appel N, Pietschmann T, Bartenschlager R (2005) Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J Virol 79:3187–3194
Upadya MH, Aweya JJ, Tan YJ (2014) Understanding the interaction of hepatitis C virus with host DEAD-box RNA helicases. World J Gastroenterol 20(11):2913–2926
Khatun M, Sur S, Steele R, Ray R, Ray RB (2021) Inhibition of long noncoding RNA linc-pint by hepatitis C virus in infected hepatocytes enhances lipogenesis. Hepatology 74(1):41–54
Marin-Bejar O, Marchese FP, Athie A, Sanchez Y, Gonzalez J, Segura V et al (2013) Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol 14:R104
Lan KH, Sheu ML, Hwang SJ, Yen SH, Chen SY, Wu JC et al (2002) HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene 21(31):4801–4811
Kao CF, Chen SY, Chen JY, Wu Lee YH (2004) Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene 23(14):2472–2483
Hoenen T, Groseth A, Feldmann H (2019) Therapeutic strategies to target the Ebola virus life cycle. Nat Rev Microbiol 17(10):593–606
Ilinykh PA, Tigabu B, Ivanov A, Ammosova T, Obukhov Y, Garron T et al (2014) Role of protein phosphatase 1 in dephosphorylation of Ebola virus VP30 protein and its targeting for the inhibition of viral transcription. J Biol Chem 289:22723–22738
Lier C, Becker S, Biedenkopf N (2017) Dynamic phosphorylation of Ebola virus VP30 in NP-induced inclusion bodies. Virology 512:39–47
Biedenkopf N, Lier C, Becker S (2016) Dynamic phosphorylation of VP30 is essential for Ebola virus life cycle. J Virol 90:4914–4925
Takamatsu Y, Krähling V, Kolesnikova L, Halwe S, Lier C, Baumeister S et al (2020) Serine-arginine protein kinase 1 regulates Ebola virus transcription. MBio 11(1):e02565-e2619
Kirchdoerfer RN, Moyer CL, Abelson DM, Saphire EO (2016) The Ebola virus VP30-NP interaction is a regulator of viral RNA synthesis. PLoS Pathog 12:e1005937
Biedenkopf N, Hartlieb B, Hoenen T, Becker S (2013) Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex: impact on viral transcription and replication. J Biol Chem 288:11165–11174
Karn J, Stoltzfus CM (2012) Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb Perspect Med 2(2):a006916
Mahiet C, Swanson CM (2016) Control of HIV-1 gene expression by SR proteins. Biochem Soc Trans 44(5):1417–1425
Huang TS, Nilsson CE, Punga T, Akusjarvi G (2002) Functional inactivation of the SR family of splicing factors during a vaccinia virus infection. EMBO Rep 3(11):1088–1093
Jacquenet S, Decimo D, Muriaux D, Darlix JL (2005) Dual effect of the SR proteins ASF/SF2, SC35 and 9G8 on HIV-1 RNA splicing and virion production. Retrovirology 2(1):33
Fukuhara T, Hosoya T, Shimizu S, Sumi K, Oshiro T, Yoshinaka Y et al (2006) Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc Natl Acad Sci USA 103(30):11329–11333
Dahal S, Clayton K, Been T, Fernet-Brochu R, Ocando AV, Balachandran A et al (2022) Opposing roles of CLK SR kinases in controlling HIV-1 gene expression and latency. Retrovirology 19(1):18
Lapek JD Jr, Lewinski MK, Wozniak JM, Guatelli J, Gonzalez DJ (2017) Quantitative temporal viromics of an inducible HIV-1 model yields insight to global host targets and phospho-dynamics associated with protein Vpr. Mol Cell Proteomics 16(8):1447–1461
Sommer G, Heise T (2008) Posttranscriptional control of HBV gene expression. Front Biosci 13:5533–5547
Selzer L, Zlotnick A (2015) Assembly and release of hepatitis B virus. Cold Spring Harb Perspect Med 5:a021394
Lan YT, Li J, Liao WY, Ou JH (1998) Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259:342–348
LiaoW OuJH (1995) Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J Virol 69:1025–1029
Kann M, Gerlich WH (1994) Effect of core protein phosphorylation by protein kinase C on encapsidation of RNA within core particles of hepatitis B virus. J Virol 68:7993–8000
Daub H, Blencke S, Habenberger P, Kurtenbach A, Dennenmoser J, Wissing J et al (2002) Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J Virol 76:8124–8137
Chen C, Wang JC, Zlotnick A (2011) A kinase chaperones hepatitis B virus capsid assembly and captures capsid dynamics in vitro. PLoS Pathog 7:e1002388
Heger-Stevic J, Zimmermann P, Lecoq L, Böttcher B, Nassal M (2018) Hepatitis B virus core protein phosphorylation: identification of the SRPK1 target sites and impact of their occupancy on RNA binding and capsid structure. PLoS Pathog 14(12):e1007488
Kim J, Wu J (2014) A theoretical study of SRPK interaction with the flexible domains of hepatitis B capsids. Biophys J 107(6):1453–1461
Lewellyn EB, Loeb DD (2011) Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PLoS ONE 6:e17202
Zheng Y, Fu XD, Ou JH (2005) Suppression of hepatitis B virus replication by SRPK1 and SRPK2 via a pathway independent of the phosphorylation of the viral core protein. Virology 342:150–158
Melegari M, Wolf SK, Schneider RJ (2005) Hepatitis B virus DNA replication is coordinated by core protein serine phosphorylation and HBx expression. J Virol 79(15):9810–9820
Chabrolles H, Auclair H, Vegna S, Lahlali T, Pons C, Michelet M et al (2020) Hepatitis B virus Core protein nuclear interactome identifies SRSF10 as a host RNA-binding protein restricting HBV RNA production. PLoS Pathog 16:e1008593
Köck J, Rösler C, Zhang JJ, Blum HE, Nassal M, Thoma C (2010) Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog 6(9):e1001082
Bergmann TK, Stage TB, Stenvang J, Christophersen P, Jacobsen TA, Roest NL et al (2020) Four phase 1 trials to evaluate the safety and pharmacokinetic profile of single and repeated dosing of SCO-101 in adult male and female volunteers. Basic Clin Pharmacol Toxicol 127:329–337
Schröder M, Leiendecker M, Grädler U, Braun J, Blum A, Wanior M et al (2023) MSC-1186, a highly selective pan-SRPK inhibitor based on an exceptionally decorated benzimidazole-pyrimidine core. J Med Chem 66(1):837–854
Batson J, Toop HD, Redondo C, Babaei-Jadidi R, Chaikuad A, Wearmouth SF et al (2017) Development of potent, selective SRPK1 inhibitors as potential topical therapeutics for neovascular eye disease. ACS Chem Biol 12(3):825–832
Hatcher JM, Wu G, Zeng C, Zhu J, Meng F, Patel S et al (2018) SRPKIN-1: a covalent SRPK1/2 inhibitor that potently converts VEGF from pro-angiogenic to anti-angiogenic isoform. Cell Chem Biol 25(4):460-470.e6
Siqueira RP, Barbosa Éde A, Polêto MD, Righetto GL, Seraphim TV, Salgado RL et al (2015) Potential antileukemia effect and structural analyses of SRPK inhibition by N-(2-(piperidin-1-yl)-5-(trifluoromethyl)phenyl)isonicotinamide (SRPIN340). PLoS ONE 10(8):e0134882
Gammons MV, Fedorov O, Ivison D, Du C, Clark T, Hopkins C et al (2013) Topical antiangiogenic SRPK1 inhibitors reduce choroidal neovascularization in rodent models of exudative AMD. Invest Ophthalmol Vis Sci 54(9):6052–6062
Gammons MV, Lucas R, Dean R, Coupland SE, Oltean S, Bates DO (2014) Targeting SRPK1 to control VEGF-mediated tumour angiogenesis in metastatic melanoma. Br J Cancer 111(3):477–485
Wodi C, Belali T, Morse R, Porazinski S, Ladomery M (2023) SPHINX-based combination therapy as a potential novel treatment strategy for acute myeloid leukaemia. Br J Biomed Sci 80:11041
Kurimchak AM, Kumar V, Herrera-Montávez C, Johnson KJ, Srivastava N, Davarajan K et al (2020) Kinome profiling of primary endometrial tumors using multiplexed inhibitor beads and mass spectrometry identifies SRPK1 as candidate therapeutic target. Mol Cell Proteomics 19:2068–2089
Montrone M, Catino A, Palmieri VO, Longo V, Galetta D (2020) Favourable outcome of coronavirus disease 2019 in a patient with anaplastic lymphoma kinase-positive non-small-cell lung cancer receiving alectinib. Eur J Cancer 138:109–112
Leonetti A, Facchinetti F, Zielli T, Brianti E, Tiseo M (2020) COVID-19 in lung cancer patients receiving ALK/ROS1 inhibitors. Eur J Cancer 132:122–124
Peng Y, Du N, Lei Y, Dorje S, Qi J, Luo T et al (2020) Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. EMBO J 39:e105938
Lin SY, Liu CL, Chang YM, Zhao J, Perlman S, Hou MH (2014) Structural basis for the identification of the N-terminal domain of coronavirus nucleocapsid protein as an antiviral target. J Med Chem 57:2247–2257
Chang CK, Jeyachandran S, Hu NJ, Liu CL, Lin SY, Wang YS et al (2016) Structure-based virtual screening and experimental validation of the discovery of inhibitors targeted towards the human coronavirus nucleocapsid protein. Mol Biosyst 12:59–66
Lin SM, Lin SC, Hsu JN, Chang CK, Chien CM, Wang YS et al (2020) Structure-based stabilization of non-native protein–protein interactions of coronavirus nucleocapsid proteins in antiviral drug design. J Med Chem 63(6):3131–3141
Hayouka Z, Rosenbluh J, Levin A, Loya S, Lebendiker M, Veprintsev D et al (2007) Inhibiting HIV-1 integrase by shifting its oligomerization equilibrium. Proc Natl Acad Sci USA 104:8316–8321
Kota S, Coito C, Mousseau G, Lavergne JP, Strosberg AD (2009) Peptide inhibitors of hepatitis C virus core oligomerization and virus production. J Gen Virol 90(Pt 6):1319–1328
Viswanathan U, Mani N, Hu Z, Ban H, Du Y, Hu J et al (2020) Targeting the multifunctional HBV core protein as a potential cure for chronic hepatitis B. Antiviral Res 182:104917
Gane E, Yuen MF, Bo Q, Schwabe C, Tanwandee T, Das S et al (2019) RO7049389, a core protein allosteric modulator, demonstrates robust decline in HBV DNA and HBV RNA in chronic HBV infected patients. J Hepatol 70:e491
Lenz O, Verbinnen T, Hodari M, Talloen W, Vandenbossche JJ, Shukla U et al (2019) HBcrAg decline in JNJ-56136379-treated chronic hepatitis b patients: results of a phase 1 study. J Hepatol 70(SUPPL 1):e473–e474
Basler CF, Krogan NJ, Leung DW, Amarasinghe GK (2019) Virus and host interactions critical for filoviral RNA synthesis as therapeutic targets. Antiviral Res 162:90–100
Modrof J, Muhlberger E, Klenk HD, Becker S (2002) Phosphorylation of VP30 impairs Ebola virus transcription. J Biol Chem 277:33099–33104
Xu W, Luthra P, Wu C, Batra J, Leung DW, Basler CF et al (2017) Ebola virus VP30 and nucleoprotein interactions modulate viral RNA synthesis. Nat Commun 8:15576
Ote I, Piette J, Sadzot-Delvaux C (2010) The Varicella–Zoster virus IE4 protein: a conserved member of the herpesviral mRNA export factors family and a potential alternative target in antiherpetic therapies. Biochem Pharmacol 80(12):1973–1980
Shanmugasundaram S, You J (2017) Targeting persistent human papillomavirus infection. Viruses 9(8):229
Moerdyk-Schauwecker M, Stein DA, Eide K, Blouch RE, Bildfell R, Iversen P et al (2009) Inhibition of HSV-1 ocular infection with morpholino oligomers targeting ICP0 and ICP27. Antiviral Res 84(2):131–141
Shkreta L, Blanchette M, Toutant J, Wilhelm E, Bell B, Story BA et al (2017) Modulation of the splicing regulatory function of SRSF10 by a novel compound that impairs HIV-1 replication. Nucleic Acids Res 45:4051–4067
Cheung PK, Horhant D, Bandy LE, Zamiri M, Rabea SM, Karagiosov SK et al (2016) A parallel synthesis approach to the identification of novel diheteroarylamide-based compounds blocking HIV replication: potential inhibitors of HIV-1 pre-mRNA alternative splicing. J Med Chem 59:1869–1879
Anderson ES, Lin CH, Xiao X, Stoilov P, Burge CB, Black DL (2012) The cardiotonic steroid digitoxin regulates alternative splicing through depletion of the splicing factors SRSF3 and TRA2B. RNA 18(5):1041–1049
Wong RW, Balachandran A, Ostrowski MA, Cochrane A (2013) Digoxin suppresses HIV-1 replication by altering viral RNA processing. PLoS Pathog 9(3):e1003241
Li Q, Jiang Z, Ren S, Guo H, Song Z, Chen S et al (2022) SRSF5-mediated alternative splicing of m gene is essential for influenza a virus replication: a host-directed target against influenza virus. Adv Sci (Weinh) 9(34):e2203088
Lu JW, Chen YC, Huang CK, Lin KC, Ho YJ (2021) Synergistic in-vitro antiviral effects of combination treatment using anidulafungin and T-1105 against Zika virus infection. Antiviral Res 195:105188
Shen S, Zhang Y, Yin Z, Zhu Q, Zhang J, Wang T et al (2022) Antiviral activity and mechanism of the antifungal drug, anidulafungin, suggesting its potential to promote treatment of viral diseases. BMC Med 20(1):359
Zhang Y, Wang M, Meng F, Yang M, Chen Y, Guo X et al (2022) A novel SRSF3 inhibitor, SFI003, exerts anticancer activity against colorectal cancer by modulating the SRSF3/DHCR24/ROS axis. Cell Death Discov 8(1):238
Wan L, Yu W, Shen E, Sun W, Liu Y, Kong J et al (2019) SRSF6-regulated alternative splicing that promotes tumour progression offers a therapy target for colorectal cancer. Gut 68:118–129
Awad IE, Abu-Saleh AAA, Sharma S, Yadav A, Poirier RA (2022) High-throughput virtual screening of drug databanks for potential inhibitors of SARS-CoV-2 spike glycoprotein. J Biomol Struct Dyn 40(5):2099–2112
Moreira GA, Caetano MMM, do Vale JA, de Paiva JC, Gonçalves VHS, Almeida AA et al (2022) The SRPK inhibitor N-(2-(piperidin-1-yl)-5-(trifluoromethyl)phenyl) isonicotinamide (SRPIN340) increases the immune response against metastatic melanoma in mice. Biochem Pharmacol 203:115161
Fortes P, Lamond AI, Ortín J (1995) Influenza virus NS1 protein alters the subnuclear localization of cellular splicing components. J Gen Virol 76(Pt 4):1001–1007
Zhao L, Li Y, Zhao Y, Liu Q, Lu Y, Ping J (2022) SRSF3 facilitates replication of influenza A virus via binding and promoting the transport of viral mRNA. Vet Microbiol 266:109343
Artarini A, Meyer M, Shin YJ, Huber K, Hilz N, Bracher F et al (2019) Regulation of influenza A virus mRNA splicing by CLK1. Antivir Res 168:187–196
Yomoda J, Muraki M, Kataoka N, Hosoya T, Suzuki M, Hagiwara M et al (2008) Combination of Clk family kinase and SRp75 modulates alternative splicing of Adenovirus E1A. Genes Cells 13:233–244
Wong R, Balachandran A, Mao AY, Dobson W, Gray-Owen S, Cochrane A (2011) Differential effect of CLK SR kinases on HIV-1 gene expression: potential novel targets for therapy. Retrovirology 8:47
Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D et al (2010) Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463:818–822
Zheng X, Peng Q, Wang L, Zhang X, Huang L, Wang J et al (2020) Serine/arginine-rich splicing factors: the bridge linking alternative splicing and cancer. Int J Biol Sci 16(13):2442–2453
Funding
This work was supported by the National Natural Science Foundation of China (82072274), and the Basic and Applied Basic Research Foundation of Guangdong Province (2021B1515120088).
Author information
Authors and Affiliations
Contributions
KZ conceptualization, resources, writing—review & editing, funding acquisition. ZR, YW conceptualization, Funding acquisition. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zheng, K., Ren, Z. & Wang, Y. Serine-arginine protein kinases and their targets in viral infection and their inhibition. Cell. Mol. Life Sci. 80, 153 (2023). https://doi.org/10.1007/s00018-023-04808-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-04808-6