
Abstract
Objective
A 1:1 ratio of fatty acid (FA)–albumin complex was chosen to mimic physiological conditions, and the effects of FA–bovine serum albumin (BSA) complexes were tested in lipopolysaccharide (LPS)-stimulated RAW264.7 macrophage cells.
Methods
Nitric oxide (NO) and various proteins/factors in RAW264.7 cells were quantified as follows: NO by the Griess assay; prostaglandin (PG) E2, interleukin (IL)-6 and tumor necrosis factor (TNF)-α by ELISA; inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2 by Western blotting; and NF-κB and CD14/TLR4 by Western blotting or flow cytometry.
Results
BSA- or FA–BSA-treated RAW264.7 cells without LPS stimulation did not show any significant changes in NO or the tested proteins/factors and thus did not have any pro-inflammatory responses. Pre-treatment with unsaturated FA–BSA complexes significantly decreased the production of LPS-induced NO, PGE2, IL-6 and TNF-α, the expression of iNOS, COX-2 and CD14, IκB degradation and NF-κB translocation. On the contrary, pre-treatment with saturated FA–BSA complexes enhanced these LPS-induced pro-inflammatory factors and the subsequent responses.
Conclusions
We concluded that unsaturated FA–BSA complexes, but not saturated FA–BSA complexes, exert an inhibitory effect on the LPS-induced pro-inflammatory response and that this effect may be partially mediated through suppression of the NF-κB signaling pathway. We suggest that an increase of unsaturated FA–BSA complexes may enhance the host’s defense against bacterial infection.
Similar content being viewed by others


Introduction
It has been well documented that certain dietary fatty acids (FAs) can differentially modulate immune responses [1–5]. In plasma, long-chain fatty acids are strongly bound to circulating albumin [6, 7], and FAs form high-affinity complexes with albumin to facilitate their solubility and transport in aqueous solution [8]. Albumin is composed of a single polypeptide chain that folds to form three or four spherical units. The strong FA-binding sites are located in crevices between these spherical regions. The binding sites are pliable, and their configurations can adapt to fit the incoming FAs [7].
During inflammation, macrophages are involved in pathogen killing, in clearing up cellular and tissue debris, and in tissue repair [2]. Macrophage activity is induced by a bacterial endotoxin (lipopolysaccharide; LPS) trigger [9]. The binding of LPS to its receptor complex (CD14/TLR4) leads to NF-κB activation and induces a pro-inflammatory response, such as the release of nitric oxide (NO), prostaglandin E2 (PGE2), interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) and the production of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) [10–13]. In unstimulated cells, NF-κB remains largely inactive in the cytoplasm as an IκB–NF-κB complex. After LPS stimulation, the cytosolic IκB is phosphorylated by IκB kinase (IKK). This phosphorylation of IκB causes it to dissociate from the IκB–NF-κB complex, allowing NF-κB to enter the nucleus [14, 15]. NF-κB then binds the target DNA elements and thus activates the transcription of pro-inflammatory genes, such as iNOS, COX-2, TNF-α and IL-6 [16–18].
Under physiological conditions, the average ratio of FA to albumin is 1:1 [19, 20]. Although different binding ratios of FA–bovine serum albumin (BSA) have been reported in a few studies on modulating inflammatory responses in various cell types [21–24], the 1:1 FA–BSA complex affects the inflammatory response, and its underlying mechanism is poorly understood. In this study, we investigated the effects of and the signal transduction pathways involving four major fatty acids—palmitic acid (PA), stearic acid (SA), oleic acid (OA) and linoleic acid (LA)—on inflammatory responses to LPS in RAW264.7 cells via the 1:1 FA–BSA complex.
Materials and methods
Chemicals and cell culture
BSA (FA-free), FA sodium salt (sodium stearate, sodium palmitate, sodium oleate, linoleic acid sodium salt) and Griess reagent (sulfanilamide, phosphoric acid and N-(1-naphthyl)ethylenediamine dihydrochloride) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies against iNOS, COX-2, IκBα and p65 and an HRP-linked goat anti-rabbit IgG secondary antibody were purchased from Cell Signaling Technology (Beverly, MA, USA). Primary antibodies against CD14 and TLR4 and an HRP-linked donkey anti-goat IgG secondary antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). A specific antibody for β-actin was purchased from Biovision (Mountain View, CA, USA). ATCC (American Type Culture Collection) murine RAW264.7 cells were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum and 100 μg/ml penicillin–streptomycin at 37°C in a humidified 5% CO2 incubator. All other culture reagents and supplies were purchased from Gibco (Invitrogen, Rockville, MD, USA).
Preparation of FA–BSA complexes
The four FAs bound with BSA (FA-free) at a 1:1 molar ratio were prepared according to the method of Mahoney et al. (1977) with modifications. Briefly, defatted BSA (1.056 g) was dissolved in Ca2+–Mg2+-free phosphate buffered saline (PBS) in a screw-capped tube. After the BSA was dissolved, the pH of the solution was adjusted to 7.2 with 1N NaOH. Salts (16 μmol) of the four different FAs were then mixed into this defatted BSA solution. After the salt was entirely dissolved, the volume of the solution was made up to 5.0 ml with PBS. This yielded a 3.2 mM FA–BSA stock solution, which was further diluted with PBS to yield a 0.32 mM working solution. The FA–BSA working solutions were membrane-filtered and then added to the culture media to a final concentration of 32 μM.
Determination of nitric oxide (NO) production
The amount of NO formation in the culture medium was estimated from the accumulation of the stable NO metabolite, nitrite, by the Griess assay [25]. Briefly, cells (1.5 × 105/ml per well in a 24-well plate) were pre-treated with 32 μM of BSA, saturated FA (SFA: PA, SA)–BSA or unsaturated FA (UFA: OA, LA)–BSA for 12 h and then stimulated with or without 1 μg/ml LPS for 24 h. The culture medium was then collected and centrifuged for 5 min at 1000g. After centrifugation, 70 μl of supernatant was transferred to a new 96-well plate, and an equivalent amount of Griess reagent [1% sulfanilamide in 5% phosphoric acid and 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride in water] was added. The optical density (OD) of the mixture was measured using a PARADIGM Detection Platform ELISA plate reader (Beckman Coulter, CA, USA) at 540 nm.
Measurement of PGE2 production
Cells (1.5 × 105/ml per well in a 24-well plate) were pre-treated with 32 μM of BSA, SFA (PA, SA)–BSA or UFA (OA, LA)–BSA for 12 h and then stimulated with or without 1 μg/ml LPS for 24 h. The culture medium was collected and assayed with a PGE2 EIA kit (Cayman, Chemicals, Ann Arbor, MI, USA). Culture medium was incubated in a goat anti-mouse IgG-coated plate with acetylcholinesterase linked to PGE2 and PGE2 monoclonal antibody for 18 h at 4°C. The plate was then emptied and rinsed with wash buffer five times. Two hundred microliters of substrate reagent was added to each well and incubated for 1 h at 37°C. Finally, the developed plate was read at 405 nm using a PARADIGM Detection Platform ELISA plate reader. The concentration of PGE2 within each sample was determined according to the standard curve.
Detection of cytokine production
Cells (1.5 × 105/ml per well in a 24-well plate) were pre-treated with 32 μM of BSA, SFA (PA, SA)–BSA or UFA (OA, LA)–BSA for 12 h and then stimulated with or without 1 μg/ml LPS for 24 h. Following stimulation, the culture medium was collected and centrifuged at 1000g for 5 min to obtain cell-free supernatant. Supernatants were stored at −80°C before being assessed for various cytokines. The concentrations of TNF-α and IL-6 in the supernatants were measured by commercial DuoSet ELISA kits (R&D Systems Europe, Abingdon, Oxon, UK) according to the manufacturer’s instructions.
Determination of iNOS, COX-2, CD14 and TLR4 expression
Cells (2 × 106/ml in a 6-cm dish) were pre-treated with 32 μM of BSA, SFA (PA, SA)–BSA or UFA (OA, LA)–BSA for 12 h and then stimulated with or without 1 μg/ml LPS for 6 h (for expression of CD14 and TLR4) or 24 h (for expression of iNOS and COX-2). Cells were collected and washed twice in PBS and were then lysed in ice-cold lysis buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin) for 30 min on ice. The lysate was centrifuged at 13000g for 15 min at 4°C. The protein content of the supernatant was measured by Bradford assay using a PARADIGM Detection Platform ELISA plate reader. Cell lysates were separated by 8% SDS-PAGE and electrophoretically transferred onto polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA, USA). After several washes, membranes were blocked with 5% skim milk in TBST (Tris-buffered saline containing 0.1% Tween-20) for 1 h at room temperature and incubated with the different primary antibodies (iNOS, COX-2, CD14 and TLR4 at 1:1000 dilution) at 4°C overnight. Membranes were then washed three times with TBST and probed with horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000) for 1 h at room temperature. After washing three times in TBST, bound antibody was visualized using ECL Western Blotting Reagent (PerkinElmer, Boston, MA, USA), and the chemiluminescence was detected using Fuji Medical X-ray film (Tokyo, Japan).
Determination of IκBα and p65 expression
After pre-treatment for 12 h with 32 μM of BSA, SFA (PA, SA)–BSA or UFA (OA, LA)–BSA and with or without LPS (1 μg/ml) stimulation for 1 h, cells were subsequently detached by gentle scraping and resuspended in ice-cold PBS. Nuclear and cytoplasmic extracts were isolated using the NE–PER Nuclear and Cytoplasmic Extraction Reagents Kit (Pierce, Rockford, IL, USA) according to the manufacturer’s instruction. The extracts were stored at −80°C until further Western blot analyses. The cytoplasmic extracts were detected with IκBα and p65 antibodies (1:5000). Nuclear extracts were detected with p65 antibody alone.
Determination of cell surface CD14 and TLR4 expression
The expression levels of CD14 and TLR4 on the surfaces of RAW264.7 macrophages were determined by flow cytometry assay. Cells (6 × 105/ml) were pre-treated with 32 μM of BSA, SFA (PA, SA)–BSA or UFA (OA, LA)–BSA for 12 h and stimulated with or without 1 μg/ml LPS for 6 h. Cells were then stained with FITC-conjugated rat anti-mouse CD14 antibody and PE-conjugated mouse anti-mouse TLR4 antibody (eBioscience, San Diego, CA, USA) for 40 min on ice. FITC-conjugated rat IgG2a (BD Biosciences) and PE-conjugated mouse IgG1 (Santa Cruz) were used as isotype controls. After washing with PBS, cell fluorescence was analyzed using a FACScan flow cytometer (Becton–Dickinson, Mountain View, CA, USA).
Statistical analysis
All data were expressed as means ± SD. Differences between the control group and experimental groups were compared by analysis of variance (ANOVA) and the post-hoc Tukey Honestly Significant Difference (HSD) test using SPSS 11.5 software for Windows (USA). P values <0.05 were considered statistically significant.
Results
Effects of SFA–BSA and UFA–BSA complexes on NO production and iNOS expression in LPS-stimulated RAW264.7 cells
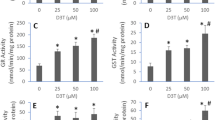
NO is recognized as a mediator and regulator of inflammatory responses [26]. To investigate whether NO formation may be modulated by SFA–BSA or UFA–BSA complexes in LPS-stimulated RAW264.7 cells, NO production was quantified by the Griess assay. As shown in Fig. 1a, low NO production was observed in the cells after treatment with BSA, PA–BSA, SA–BSA, OA–BSA or LA–BSA without LPS. A dramatic increase in NO production was seen in cells following LPS stimulation for 24 h. However, this increase in NO production was significantly suppressed in LPS-stimulated cells by pre-treatment with UFA–BSA (p < 0.05), but not with SFA–BSA. It is known that NO is synthesized by iNOS in macrophages against microorganisms and tumor cells [27, 28]. Western blotting illustrated that the expression of iNOS was detected only in LPS-stimulated cells and not in cells not exposed to LPS (Figs. 1b, c). In addition, the expression of iNOS in the cells was significantly reduced by LA–BSA pre-treatment (p < 0.01) but was significantly increased by SA–BSA pre-treatment (p < 0.05).

Effects of SFA–BSA and UFA–BSA on NO production and iNOS expression in LPS-stimulated RAW264.7 cells. Cells were pre-treated with 32 μM of BSA, SFA (PA, SA)–BSA or UFA (OA, LA)–BSA for 12 h and then stimulated with or without LPS (1 μg/ml) for 24 h. a Nitrite in the medium was measured by the addition of Griess reagent. The data represent three separate measurements. *p < 0.05 compared to the BSA + LPS group (10.6 μM ± 0.3). b The whole-cell lysates were separated on 8% SDS-PAGE. The level of iNOS was analyzed by Western blotting. c Total protein loaded in each lane was normalized with reference to actin, and the relative levels of iNOS expression between experimental groups were measured. The data are presented as means ± SD of three independent experiments. *p < 0.05 and **p < 0.01 compared to the BSA + LPS group (100 ± 0.006%)
Effects of SFA–BSA and UFA–BSA complexes on PGE2 production and COX-2 expression in LPS-stimulated RAW264.7 cells
PGE2 is a crucial mediator of the inflammatory process [29]. An increase in PGE production is found in macrophages following LPS treatment [30]. The amount of PGE2 release in the culture medium was measured by ELISA assay to determine the effect of different FA–BSA complexes on PGE2 production in LPS-stimulated RAW264.7 cells. Compared with the BSA-only treatment group, LPS (1 μg/ml) treatment induced a substantial production of PGE2 in RAW264.7 cells. Pre-treatment with UFA–BSA, especially OA–BSA, significantly decreased the production of PGE2 in macrophages stimulated with LPS (p < 0.05). However, SFA–BSA pre-treatment, especially SA–BSA, facilitated the production of PGE2 in LPS-stimulated macrophages (p < 0.01) (Fig. 2a).

Effects of SFA–BSA and UFA–BSA on PGE2 production and COX-2 expression in LPS-stimulated RAW264.7 cells. Cells were pre-treated with 32 μM of BSA, SFA (PA, SA)–BSA or UFA (OA, LA)–BSA for 12 h and then stimulated with or without LPS (1 μg/ml) for 24 h. a PGE2 EIA assay (BSA + LPS group: 1.76 × 105 ± 0.124 × 105 pg/ml). b, c Western blot analysis of COX-2 expression was performed as described in “Materials and methods”. Each column represents the means ± SD from three independent experiments. *p < 0.05 and **p < 0.01 indicated significant differences from the BSA + LPS group. Note in panel a that in cells treated with BSA, PA–BSA, SA–BSA, OA–BSA and LA–BSA, PGE2 production is not detectable by the PGE2 EIA kit
COX-2 is highly induced at inflammatory sites in animals and in patients with inflammatory diseases [31, 32] and is responsible for pro-inflammatory PG formation. Thus, in this study, we assessed the level of COX-2 protein expression by Western blotting. Our results revealed that a significant increase in COX-2 protein was observed in LPS-stimulated RAW264.7 cells, but not in cells without LPS treatment (Fig. 2b). However, pre-treatment with UFA–BSA attenuated COX-2 expression in LPS-stimulated cells (p < 0.01). In contrast, COX-2 expression was increased in LPS-stimulated cells with SFA–BSA pre-treatment (Fig. 2c).
Effects of SFA–BSA and UFA–BSA complexes on IL-6 and TNF-α production in LPS-stimulated RAW264.7 cells
To determine the effects of SFA–BSA and UFA–BSA complexes on the release of cytokines induced by LPS, IL-6 and TNF-α were measured by ELISA. Figures 3a, b show that LPS-induced IL-6 and TNF-α production was significantly decreased by UFA–BSA pre-treatment but not by SFA–BSA pre-treatment in RAW264.7 cells. In addition, we failed to detect IL-6 expression in the cells exposed to SFA–BSA or UFA–BSA alone without LPS stimulation. Taken together, these data suggest that UFA–BSA, but not SFA–BSA, has an anti-inflammatory effect by decreasing pro-inflammatory cytokine production.

Effects of SFA–BSA and UFA–BSA on IL-6 and TNF-α expression in LPS-stimulated RAW264.7 cells. After pre-treatment with 32 μM of BSA, SFA (PA, SA)–BSA or UFA (OA, LA)–BSA for 12 h, cells were then stimulated with or without LPS (1 μg/ml) for 24 h. The amounts of a IL-6 (BSA + LPS group: 1817.7 pg/ml ± 187.4) or b TNF-α (BSA + LPS group: 12883.6 pg/ml ± 150.7) released within the medium were measured by ELISA. Data were expressed as means ± SD (n = 3). *p < 0.05 or **p < 0.01 compared to the BSA+LPS group. Note in panel a that in cells treated with BSA, PA–BSA, SA–BSA, OA–BSA and LA–BSA, IL-6 production is not detectable by the IL-6 ELISA kit
Effects of SFA–BSA and UFA–BSA complexes on NF-κB nuclear translocation and IκBα degradation in LPS-stimulated RAW264.7 cells
The NF-κB pathway is activated in cells challenged by LPS and is involved in the transcriptional activation of the response genes iNOS and COX-2 [33, 34]. To investigate the possible mechanism that SFA–BSA and UFA–BSA complexes use to modulate LPS-induced iNOS and COX-2 protein expression, intracellular NF-κB signaling was further examined. Western blot analyses showed that p65 expression in the nuclear fraction was increased after 1 h of LPS stimulation compared with the group without LPS exposure (Figs. 4a, b). The expression of LPS-induced p65 was reduced in cells after LA–BSA or OA–BSA pre-treatment. However, the expression of LPS-induced p65 was enhanced in cells pre-treated with PA–BSA or SA–BSA. Consistent with the above results, the cytosolic levels of the p65 subunit of NF-κB in LPS-stimulated cells pre-treated with UFA–BSA complexes were higher, although not statistically significant, than those in LPS-stimulated cells pre-treated with SFA–BSA complexes (Figs. 4c, d).

Effects of SFA–BSA and UFA–BSA on IκBα degradation and translocation of the p65 subunit of NF-κB in LPS-stimulated RAW264.7 cells. Cells were pre-treated with 32 μM of BSA, SFA (PA, SA)–BSA or UFA (OA, LA)–BSA for 12 h and were then stimulated with or without LPS (1 μg/ml) for 1 h. The cytosolic and nuclear fractions were separated. The lysates were then subjected to 8% SDS-PAGE followed by Western blot analysis. a Western blot analysis of the p65 subunit of NF-κB in the nuclear fraction. c Western blot analyses with specific antibodies for IκBα and the p65 subunit of NF-κB in the cytoplasmic fraction. b, d, e: each column represents the means ± SD from three independent experiments
Because cytosolic IκBα inhibits NF-κB transcriptional activity, we examined the effect of SFA–BSA and UFA–BSA on the level of IκBα in LPS-stimulated RAW264.7 cells. As shown in Figs. 4c, e, the expression of IκBα had no significant change in RAW264.7 macrophages following BSA or FA–BSA treatment. IκBα expression in LPS-stimulated cells pre-treated with UFA–BSA was higher, although not statistically significant, than in LPS-stimulated cells pre-treated with SFA–BSA. These data confirm that both the degradation of IκBα and the translocation of the p65 subunit of NF-κB induced by LPS in RAW264.7 cells were attenuated by UFA–BSA complexes.
Effects of SFA–BSA and UFA–BSA complexes on CD14 and TLR4 expression in LPS-stimulated RAW264.7 cells
Because the CD14/TLR4 complex plays a vital role in initiating LPS signaling during inflammation [35], we further examined the expressions of CD14 and TLR4 modulated by SFA–BSA and UFA–BSA in LPS-induced RAW264.7 cells. Our results showed that the expression of CD14 was weakly detected in RAW264.7 cells without exposure to LPS. After LPS stimulation, CD14 expression was significantly up-regulated and detectable (p < 0.001) (Fig. 5a, d). Results from flow cytometry revealed that no significant differences in the fluorescence intensity of CD14 were found in the cells treated with BSA, SFA–BSA or UFA–BSA. A marked increase in CD14 fluorescence intensity was observed in cells following a 6-h treatment with LPS (p < 0.001). Only a slight decrease in CD14 fluorescence intensity was observed in LPS-stimulated cells following pre-treatment with LA–BSA (Fig. 5b, d). The effect of SFA–BSA and UFA–BSA on TLR4 expression was subsequently investigated. Western blotting data demonstrated that TLR4 expression was very low and hard to detect (Fig. 5a). Similar results were obtained from flow cytometry (Fig. 5c).

Effects of SFA–BSA and UFA–BSA on CD14 and TLR4 expression in LPS-stimulated RAW264.7 cells. Cells were pre-treated with 32 μM of BSA, SFA (PA, SA)–BSA or UFA (OA, LA)–BSA for 12 h and were then stimulated with or without LPS (1 μg/ml) for 6 h. a Whole-cell lysates were separated by 8% SDS-PAGE. CD14 and TLR4 levels were analyzed by Western blot. b, c FACS analysis of RAW264.7 cells labeled with b FITC-conjugated anti-CD14 antibody and c PE-conjugated anti-TLR4 antibody. In panels b and c, the thin solid black lines are isotype controls, the dotted lines indicate BSA, PA–BSA, SA–BSA, OA–BSA and LA–BSA treatment (from left to right, respectively) and bold grey lines indicate LPS stimulation after BSA or FA–BSA pre-treatment. d Quantified data from the CD14 Western blot and flow cytometry were obtained from three independent experiments and expressed as means ± SD. **p < 0.01 compared to the without-LPS group
Discussion
In this study, we found that BSA, SFA–BSA or UFA–BSA treatments could not cause pro-inflammatory responses in RAW264.7 macrophages without LPS stimulation. Pro-inflammatory responses, such as the production of NO and PGE2, the expression of iNOS and COX-2 and the release of IL6 and TNF-α, were identified in RAW264.7 cells following LPS stimulation. Previous studies have demonstrated that pro-inflammatory responses may be enhanced by most SFAs but reduced by most UFAs [22, 23, 36]. However, our data (Figs. 1–3) showed no significant changes in the production of NO, PGE2 or cytokines or the expression of iNOS or COX-2 in RAW264.7 cells treated with BSA, SFA–BSA or UFA–BSA without LPS stimulation. These results indicate that the 1:1 ratio of the FA–BSA complex was in the physiological range and did not induce any pro-inflammatory response.
Inflammation leads to the up-regulation of pro-inflammatory enzymes, such as iNOS and COX-2, and the release of numerous cytokines in affected tissues and cells [37]. iNOS is a member of the NOS protein family that can catalyze the formation of NO from l-arginine [16, 38]. It is also known that low concentrations of NO produced by iNOS in macrophages are likely to contribute to the antimicrobial activities against certain bacterial pathogens. However, high concentrations of NO and its derivatives, such as nitrogen dioxide and peroxynitrite, are involved in inflammation and carcinogenesis [39]. Ambrozova et al. (2010) [40] reported that LA can inhibit LPS-induced nitrite accumulation and suggested that UFAs may exhibit scavenging activity against NO production. Consistent with the results reported by Ambrozova et al. [41], we demonstrated that both the production of NO and the expression of iNOS by LPS were markedly reduced in RAW264.7 cells after pre-treatment with UFA–BSA (i.e., OA and LA), but not with SFA–BSA (i.e., PA and SA) (Fig. 1a, b). These results confirmed the fact that OA suppressed the expression of iNOS in LPS-stimulated BV2 microglia.
COX-2, an enzyme that catalyzes the conversion of arachidonic acid to PGEs, especially PGE2, is an inflammatory mediator inducible by LPS in macrophages [42, 43]. COX-2 is activated by various fatty acids in macrophages and the role of COX-2 in anti-inflammation and antioxidative stress are clearly reviewed in a recently study [44]. Lee et al. (2003) [5] stated that the UFAs OA and LA might exert inhibitory effects on LPS-induced COX-2 expression in macrophages. Additionally, the inhibition of LPS-induced COX-2 expression and PGE2 production were also confirmed in microglia with OA treatment [41]. In this study, we reported that UFA–BSA, especially OA, strongly inhibited COX-2 expression and subsequently reduced the production of PGE2 in LPS-stimulated RAW264.7 cells (Fig. 2a, b). Moreover, the release of IL-6 and TNF-α were decreased by 12-h pre-treatment with UFA–BSA. However, pre-treatment with SFA–BSA increased the LPS-induced release of pro-inflammatory mediators, such as IL-6 and TNF-α, in RAW264.7 cells (Fig. 3a, b). These results were consistent with a previous study which reported that LA decreased, but PA increased, IL-6 and TNF-α production in response to LPS in THP-1 (human acute monocytic leukemia cell line) cells [24].
Many inflammatory gene expressions, including iNOS, COX-2, IL-6 and TNF-α, are regulated by the transcription factor NF-κB in response to a severe inflammatory reaction [18, 45]. Generally, NF-κB is present in the cytoplasm in an inactive state, associated with an inhibitory subunit, IκB-α. However, with the phosphorylation of IκB-α by IKK, IκB-α may undergo degradation and thus can liberate NF-κB in the cytosol. This cytosolic NF-κB subsequently translocates into the nucleus and then binds the κB motifs in the promoters via its p65 subunit, leading to the expression of inflammatory genes [46, 47]. To explore whether UFA–BSA complexes have a potent inhibitory effect on NF-κB activation by LPS stimulation, the amounts of the cytosolic IκB and p65 subunits and the nuclear p65 subunit were determined separately in LPS-stimulated RAW264.7 cells. As expected, UFA–BSA pre-treatment in LPS-stimulated cells preserved more IκB and p65 subunits in the cytosol and thus blocked the translocation of p65 subunits to the nucleus (Fig. 4a, b). These results also demonstrated that UFA–BSA blocked NF-κB activation, at least in part, via the inhibition of IκB-α degradation in LPS-stimulated RAW264.7 cells (Fig. 4d, e).
LPS binds to LPS receptors, including CD14 and TLR4, before it stimulates macrophages to release pro-inflammatory cytokines [48, 49]. CD14 is a glycosylphosphatidylinositol-linked protein and transduces its signal by associating with other partners, especially TLR4 [50, 51]. It has been reported that LPS-stimulated CD14 expression and TLR-induced signaling pathways are down-regulated by ω–3 fatty acids [5, 52]. In this study, the LPS-induced CD14 expression in RAW264.7 cells was only slightly attenuated by LA–BSA pre-treatment, but was increased by SA–BSA pre-treatment (Fig. 5d). However, TLR4 expression in RAW264.7 cells, with and without LPS stimulation, was too low to determine (Fig. 5a, c). Lee et al. (2001, 2003) [53, 54] reported that the TLR4 can be activated by lauric acid (a medium-chain saturated fatty acid). However, our results obtained from the saturated fatty acids (SA and PA) are not consistent with their findings. This controversy may be due to the difference in chain length, concentration and BSA binding of the fatty acids we used. Although the total protein of TLR4 was unchanged after fatty acid treatment, we cannot rule out the possibility that TLR4 might be activated through conformation change. The involvement of TLR4 or other TLRs in the effects of SFA–BSA and UFA–BSA complexes on LPS-induced pro-inflammatory responses in RAW264.7 cells remains to be determined. Further studies using knockout and/or knockdown or overexpression of TLR4 or other TLRs will be required to address this question.
In summary, we confirmed that the pro-inflammatory responses induced by LPS in RAW264.7 cells were significantly attenuated by UFA–BSA pre-treatment, but were enhanced by SFA–BSA pre-treatment. Moreover, the modulator effect of UFA–BSA on the pro-inflammatory response induced by LPS may be partially explained by the inhibition of the NF-κB signaling pathway. In addition, a prominent increase in NO, PGE2 and cytokine production in LPS-stimulated cells with SFA–BSA pre-treatment indicated that the SFAs, rather than the UFAs, may augment the sensitivity of macrophages and may promote the pro-inflammatory response to LPS.
References
Hwang D, Rhee SH. Receptor-mediated signaling pathways: potential targets of modulation by dietary fatty acids. Am J Clin Nutr. 1999;70:545–56.
Calder PC. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am J Clin Nutr. 2006;83:1505S–19S.
Weatherill AR, Lee JY, Zhao L, Lemay DG, Youn HS, Hwang DH. Saturated and polyunsaturated fatty acids reciprocally modulate dendritic cell functions mediated through TLR4. J Immunol. 2005;174:5390–7.
Baldie G, Kaimakamis D, Rotondo D. Fatty acid modulation of cytokine release from human monocytic cells. Biochim Biophys Acta. 1993;1179:125–33.
Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, et al. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res. 2003;44:479–86.
Elmadhoun BM, Wang GQ, Templeton JF, Burczynski FJ. Binding of [3H]palmitate to BSA. Am J Physiol. 1998;275:G638–44.
Spector AA. Fatty acid binding to plasma albumin. J Lipid Res. 1975;16:165–79.
Ulloth JE, Casiano CA, De Leon M. Palmitic and stearic fatty acids induce caspase-dependent and -independent cell death in nerve growth factor differentiated PC12 cells. J Neurochem. 2003;84:655–68.
Meng F, Lowell CA. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J Exp Med. 1997;185:1661–70.
Dobrovolskaia MA, Vogel SN. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002;4:903–14.
Tripathi S, Bruch D, Kittur DS. Ginger extract inhibits LPS induced macrophage activation and function. BMC Complem Altern Med. 2008;8:1.
Sugiyama K, Muroi M, Tanamoto K, Nishijima M, Sugita-Konishi Y. Deoxynivalenol and nivalenol inhibit lipopolysaccharide-induced nitric oxide production by mouse macrophage cells. Toxicol Lett. 2010;192:150–4.
Lopez-Bojorquez LN, Arechavaleta-Velasco F, Vadillo-Ortega F, Montes-Sanchez D, Ventura-Gallegos JL, Zentella-Dehesa A. NF-kappaB translocation and endothelial cell activation is potentiated by macrophage-released signals co-secreted with TNF-alpha and IL-1beta. Inflamm Res. 2004;53:567–75.
Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52.
Yamamoto Y, Gaynor RB. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29:72–9.
de Vera ME, Shapiro RA, Nussler AK, Mudgett JS, Simmons RL, Morris SM Jr, et al. Transcriptional regulation of human inducible nitric oxide synthase (NOS2) gene by cytokines: initial analysis of the human NOS2 promoter. Proc Natl Acad Sci USA. 1996;93:1054–9.
Makarov SS. NF-kappaB as a therapeutic target in chronic inflammation: recent advances. Mol Med Today. 2000;6:441–8.
Lee SJ, Bai SK, Lee KS, Namkoong S, Na HJ, Ha KS, et al. Astaxanthin inhibits nitric oxide production and inflammatory gene expression by suppressing I(kappa)B kinase-dependent NF-kappaB activation. Mol Cell. 2003;16:97–105.
Mahoney EM, Hamill AL, Scott WA, Cohn ZA. Response of endocytosis to altered fatty acyl composition of macrophage phospholipids. Proc Natl Acad Sci USA. 1977;74:4895–9.
Wiesenfeld PW, Babu US, O’Donnell MW. Effect of long-chain fatty acids in the culture medium on fatty acid composition of WEHI-3 and J774A.1 cells. Comp Biochem Physiol B Biochem Mol Biol. 2001;128:123–34.
Toborek M, Lee YW, Garrido R, Kaiser S, Hennig B. Unsaturated fatty acids selectively induce an inflammatory environment in human endothelial cells. Am J Clin Nutr. 2002;75:119–25.
Ajuwon KM, Spurlock ME. Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNF alpha expression in 3T3–L1 adipocytes. J Nutr. 2005;135:1841–6.
Harvey KA, Walker CL, Pavlina TM, Xu Z, Zaloga GP, Siddiqui RA. Long-chain saturated fatty acids induce pro-inflammatory responses and impact endothelial cell growth. Clin Nutr. 2010;29:492–500.
Zhao G, Etherton TD, Martin KR, Vanden Heuvel JP, Gillies PJ, West SG, et al. Anti-inflammatory effects of polyunsaturated fatty acids in THP-1 cells. Biochem Biophys Res Commun. 2005;336:909–17.
Ishihara T, Kohno K, Ushio S, Iwaki K, Ikeda M, Kurimoto M. Tryptanthrin inhibits nitric oxide and prostaglandin E(2) synthesis by murine macrophages. Eur J Pharmacol. 2000;407:197–204.
Korhonen R, Lahti A, Kankaanranta H, Moilanen E. Nitric oxide production and signaling in inflammation. Curr Drug Targets Inflamm Allergy. 2005;4:471–9.
Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, et al. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375:408–11.
Farias-Eisner R, Sherman MP, Aeberhard E, Chaudhuri G. Nitric oxide is an important mediator for tumoricidal activity in vivo. Proc Natl Acad Sci USA. 1994;91:9407–11.
Dekkers JA, Akkermans LM, Kroese AB. Effects of the inflammatory mediator prostaglandin E2 on myenteric neurons in guinea pig ileum. Am J Physiol. 1997;272:G1451–6.
Kunkel SL, Chensue SW, Phan SH. Prostaglandins as endogenous mediators of interleukin 1 production. J Immunol. 1986;136:186–92.
Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, et al. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA. 1994;91:3228–32.
Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci USA. 1994;91:12013–7.
Surh YJ, Chun KS, Cha HH, Han SS, Keum YS, Park KK, et al. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat Res. 2001;480–481:243–68.
Lappas M, Permezel M, Georgiou HM, Rice GE. Nuclear factor kappa B regulation of proinflammatory cytokines in human gestational tissues in vitro. Biol Reprod. 2002;67:668–73.
Xia Y, Yamagata K, Krukoff TL. Differential expression of the CD14/TLR4 complex and inflammatory signaling molecules following i.c.v. administration of LPS. Brain Res. 2006;1095:85–95.
Yang M, Cook ME. Dietary conjugated linoleic acid decreased cachexia, macrophage tumor necrosis factor-alpha production, and modifies splenocyte cytokines production. Exp Biol Med (Maywood). 2003;228:51–8.
Kuo YC, Lai CS, Wang JM, Badmaev V, Nagabhushanam K, Ho CT, et al. Differential inhibitory effects of inotilone on inflammatory mediators, inducible nitric oxide synthase and cyclooxygenase-2, in LPS-stimulated murine macrophage. Mol Nutr Food Res. 2009;53:1386–95.
Nathan C, Xie QW. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78:915–8.
Ohshima H, Bartsch H. Chronic infections and inflammatory processes as cancer risk factors: possible role of nitric oxide in carcinogenesis. Mutat Res. 1994;305:253–64.
Ambrozova G, Pekarova M, Lojek A. Effect of polyunsaturated fatty acids on the reactive oxygen and nitrogen species production by raw 264.7 macrophages. Eur J Nutr. 2010;49:133–9.
Oh YT, Lee JY, Lee J, Kim H, Yoon KS, Choe W, et al. Oleic acid reduces lipopolysaccharide-induced expression of iNOS and COX-2 in BV2 murine microglial cells: possible involvement of reactive oxygen species, p38 MAPK, and IKK/NF-kappaB signaling pathways. Neurosci Lett. 2009;464:93–7.
Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–73.
Peng BW, Lin JY, Zhang T. Toxoplasma gondii induces prostaglandin E2 synthesis in macrophages via signal pathways for calcium-dependent arachidonic acid production and PKC-dependent induction of cyclooxygenase-2. Parasitol Res. 2008;102:1043–50.
Luo C, Urgard E, Vooder T, Metspalu A. The role of COX-2 and Nrf2/ARE in anti-inflammation and antioxidative stress: aging and anti-aging. Med Hypotheses. 2011;77:174–8.
Srivastava SK, Ramana KV. Focus on molecules: nuclear factor-kappaB. Exp Eye Res. 2009;88:2–3.
de Martin R, Vanhove B, Cheng Q, Hofer E, Csizmadia V, Winkler H, et al. Cytokine-inducible expression in endothelial cells of an I kappa B alpha-like gene is regulated by NF kappa B. EMBO J. 1993;12:2773–9.
Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11.
Lichtman SN, Wang J, Lemasters JJ. LPS receptor CD14 participates in release of TNF-alpha in RAW 264.7 and peritoneal cells but not in Kupffer cells. Am J Physiol. 1998;275:G39–46.
Yang Z, Carter CD, Miller MS, Bochsler PN. CD14 and tissue factor expression by bacterial lipopolysaccharide-stimulated bovine alveolar macrophages in vitro. Infect Immun. 1995;63:51–6.
Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest. 2000;105:497–504.
da Silva Correia J, Soldau K, Christen U, Tobias PS, Ulevitch RJ. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. transfer from CD14 to TLR4 and MD-2. J Biol Chem. 2001;276:21129–35.
Chu AJ, Walton MA, Prasad JK, Seto A. Blockade by polyunsaturated n-3 fatty acids of endotoxin-induced monocytic tissue factor activation is mediated by the depressed receptor expression in THP-1 cells. J Surg Res. 1999;87:217–24.
Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–9.
Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, et al. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-Kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041–51.
Acknowledgments
We would like to express our sincere thanks to Dr. Ta-Hsien Chuang, School of Pharmacy, China Medical University, Taichung, Taiwan, for his critical reading and valuable discussion on this manuscript. This work was supported in part by grants (NSC98-2628-13-002-085-MY3 to KSL) from the National Science Council, Taiwan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Graham Wallace.
Rights and permissions
About this article
Cite this article
Chang, C.F., Chau, Y.P., Kung, H.N. et al. The lipopolysaccharide-induced pro-inflammatory response in RAW264.7 cells is attenuated by an unsaturated fatty acid–bovine serum albumin complex and enhanced by a saturated fatty acid–bovine serum albumin complex. Inflamm. Res. 61, 151–160 (2012). https://doi.org/10.1007/s00011-011-0399-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-011-0399-1