
Abstract
Heterotrimeric guanine nucleotide-binding proteins (G proteins), which consist of an α-, a β- and a γ-subunit, have crucial roles as molecular switches in the regulation of the downstream effector molecules of multiple G protein-coupled receptor signalling pathways, such as phospholipase C and adenylyl cyclase. According to the structural and functional similarities of their α-subunits, G proteins can be divided into four subfamilies: Gαs, Gαi/o, Gαq/11 and Gα12/13. Most of the α- and the βγ-subunits are abundantly expressed on the surface of immune cells. Recent studies have demonstrated that G proteins are a group of important immunomodulatory factors that regulate the migration, activation, survival, proliferation, differentiation and cytokine secretion of immune cells. In this review, we summarise the recent findings on the functions of G proteins in immune regulation and autoimmunity.
Similar content being viewed by others


Introduction
Mammalian cells do not exist in isolation. Thousands of molecular messages need to be received by the receptors on the cell surface, and the signals then need to be transmitted from outside to inside the cell through a chain of signalling molecules (Bockaert et al. 2002). Receptors can sometimes activate intracellular effectors more indirectly through a second messenger system, such as the G protein-mediated transmembrane signalling system. In this complex system, heterotrimeric G proteins function as critical molecular switches (Koelle 2006). The G proteins, together with G protein-coupled receptors (GPCRs), effectors and various regulators of G protein signalling (RGS), transduce the extracellular stimuli, such as hormones, chemokines, neurotransmitters, nucleotides, amino acids, sensory stimuli, biogenic amines and ions, into intercellular signals that ultimately convert these stimuli into the appropriate physiological response (Wettschureck and Offermanns 2005).
The heterotrimeric G protein consists of an α-subunit, which is a 37–42-kDa protein that contains a guanine nucleotide-binding pocket and intrinsic GTPase activity that binds and hydrolyses GTP, a 35-kDa β-subunit and an 8–11-kDa γ-subunit. The β- and γ-subunits form an indissociable complex, which is often referred to as a single entity, the βγ-subunit (Entschladen et al. 2011). When a ligand interacts with a GPCR on the surface of the cell, the Gα exchanges GTP for GDP and detaches from the Gβγ dimer. Each of the activated components then interacts with different effectors, such as adenylyl cyclase, phosphodiesterase, phospholipase C (PLC), G protein-regulated kinase and voltage-dependent Ca2+ channel, to initiate various cellular responses. The α-subunit of the heterotrimeric G protein defines the basic properties of the G protein. Based on the differences in their α-subunits, G proteins can be divided into four subfamilies: Gαs, Gαi/o, Gαq/11 and Gα12/13 (Wettschureck and Offermanns 2005). To date, only 17 different α-subunits, 5 different β-subunits and 12 different γ-subunits have been identified. Thus, a relatively small number of G proteins transmit the signals that are received by the huge number of different GPCRs (Goldsmith and Dhanasekaran 2007).
Due to its high complexity and versatility, the G protein-mediated signalling system has turned out to be the most widely used transmembrane signalling system in higher organisms (Wettschureck and Offermanns 2005). Numerous studies have focused on the function of G protein signalling in multiple tissues and organs. Some of these studies focused on the functions of this signalling pathway in the regulation of smooth and cardiac muscle contraction, platelet activation, synaptic transmission, and carbohydrate and lipid metabolism (Wettschureck and Offermanns 2005). A large number of GPCRs, including chemokine receptors, and most of the G protein subunits are widely expressed in various cell types of the immune system (Druey 2009). Through the use of knockout (KO) mice and chemical inhibitors, the functions of G proteins in the immune system have been recently studied. Increasing amounts of accumulated data have indicated that G protein signalling systems are key determinants in innate and adaptive immunity. These systems are involved in lymphocyte development, immune cell survival and migration, and tolerance induction (Table 1). In this review, we summarise the recent findings that elucidate the role of heterotrimeric G proteins in immune regulation and highlight the emerging roles of these proteins in autoimmune diseases.
G Proteins Are Required for Lymphocyte Development
The major causative factors that induce the onset and development of autoimmune diseases are a disruption in the normal development of the lymphoid tissues and an alteration in the function of lymphoid cells. The development of lymphocytes is regulated at many levels, and some G proteins have emerged as regulating factors in the commitment of precursor cells to the lymphoid lineage.
The initial study in Gαq-KO mice found that proportions of B cells, T cells, granulocytes, and monocytes from the peripheral blood, spleen, thymus, lymph nodes, Peyer’s patches, and bone marrow are not significantly different from those observed in wild-type (WT) animals (Davignon et al. 2000). However, a further analysis of this subpopulation of immune cells in Gαq-KO mice revealed that Gαq can regulate the transition between pro- and pre-B cells in the bone marrow and the development of follicular and marginal zone B cells in the spleen. The transitional 1 (T1) B cells and marginal zone B (MZB) cells of Gαq-deficient mice were largely resistant to anti-IgM-induced cell death in vitro. The abnormal survival of B cells in the transitional stage (from T1 to T2 B cells) can reduce the stringency of the negative selection of Gαq−/− B cells and allow the entry of potentially autoreactive cells into the mature MZB compartment (Meyer-Bahlburg et al. 2008; Misra et al. 2010; Thien et al. 2004).
The deletion of Gαi2 greatly augments the response of thymocytes to T-cell receptor (TCR)-mediated stimulation and results in an enhanced proliferation of double-positive (DP) thymocytes upon ligation of the TCRs. This altered response promotes positive selection and may also rescue some thymocytes from death due to their inability to bind MHC molecules (Elgbratt et al. 2007; Zhang et al. 2005). Gαi2−/− mice exhibit a significantly reduced fraction of DP thymocytes and an increased fraction of single-positive (SP) thymocytes. The absence of the Gαi2 protein also causes the arrest of thymocyte differentiation at the double-negative (DN) stage (Jin and Wu 2008). The reduced number of DP thymocytes was due to an accelerated transition from DP to SP thymocytes and a reduced transition from DN to DP thymocytes. Thus, the lower number of DP thymocytes resulted in a lower daily production by the thymus; the exact mechanisms behind the altered transition rates provided important information on thymic atrophy during colitis (Elgbratt et al. 2012). In addition, the null mutation of the Gαi2 protein resulted in disordered B-cell subpopulations in the spleen and peritoneal cavity. Gαi2−/− mice exhibited a reduced amount of MZB and T2 B cells and significantly increased numbers of follicular B cells. Gαi2 was also important for the development and recruitment of B-1a and B-1b cells from the peritoneal cavity (Dalwadi et al. 2003).
The analysis of the cells that are generated from the thymus of these mutant mice revealed that Gα13- and not Gα12-mediated signalling plays an important role in the proliferation and survival of thymocytes during development and is required for early thymopoiesis (Coffield et al. 2004). Moreover, the Gα12/Gα13 family of G proteins can also regulate MZB cell homeostasis. Although the splenic follicular structure was roughly normal, further studies of the immune cell subpopulation revealed a strong reduction of MZB cell numbers in Gα12/Gα13-double-knockout (Gα12/Gα13-DKO) mice (Rieken et al. 2006).
G Proteins Control Lymphocyte Proliferation and Survival
Lymphocyte proliferation and death must be finely regulated to maintain immune homeostasis throughout the lifetime of a mammalian organism (Hildeman et al. 2007; Xu and Shi 2007). The clonal expansion of antigen-specific lymphocytes is required for effective immune responses against invading microorganisms. Shortly after the pathogens are controlled, the expanded effector cells must be eliminated to prevent the non-adaptive accumulation of cells and ensure the return to immune homeostasis (Holtzman et al. 2000). A fine balance between the survival and the death of lymphocytes ensures an effective immune system and maintains the immunological tolerance to self (Strasser and Bouillet 2003; von Boehmer and Melchers 2010). Although lymphocyte proliferation and death are not directly regulated by G proteins, the G protein-mediated signalling system might have important modulatory roles in these immune functions.
In recent years, studies with KO mice supported the pivotal role of the Gαq-mediated signalling pathway in the regulation of lymphocyte proliferation and survival (Misra et al. 2010; Molon et al. 2005; Ngai et al. 2008, 2009; Wang et al. 2011). The G protein αq-subunit can directly inhibit PI3 K activation and prevent the activation of Akt. The PI3 K-Akt signalling pathway regulates many normal cellular processes, including cell proliferation, survival, growth, and motility processes (Browne et al. 2009; Cantrell 2002; Yanamadala et al. 2009). When chemokine receptors signal through Gαq, the activated T cells showed increased proliferation and cytokine production (Molon et al. 2005). Because Gαq−/− B cells are intrinsically defective and more resistant than WT B cells to cell death-inducing signals, such as B-cell-activating factor withdrawal and strong B-cell receptor (BCR) signals, mature Gαq−/− B cells were more fit to survive than WT B cells (Misra et al. 2010). Moreover, the level of Gαq expression can contribute to the determination of the apoptosis and survival of human peripheral blood lymphocytes (PBLs) through the upregulation of Mcl-1 and the downregulation of caspase-3 activity (Wang et al. 2011). Consistent with the findings that have shown that Gαq is involved in human T lymphocyte regulation, the targeted mutation of RGS2 in mice also lead to reduced T-cell proliferation. RGS2 interacts with Gαq/11 and accelerates the GTPase activity of the α-subunit, which negatively regulates G protein-coupled receptor signalling (Oliveira-Dos-Santos et al. 2000).
Despite the critical role of Gαq in lymphocyte proliferation and survival, studies with Gαi2-deficient mice have recently demonstrated that the induction of the Gαi2-mediated signalling pathways is sufficient to negatively regulate T-cell proliferation (Gotlind et al. 2011; Hornquist et al. 1997; Jiang et al. 1997; Zhang et al. 2005). Gαi2−/− peripheral T cells display a hyperimmune response, which is characterised by an enhanced proliferation and production of inflammatory cytokines in response to stimulation with various mitogens (Hornquist et al. 1997; Huang et al. 2003). Interestingly, Gαi2−/− T and B lymphocytes exhibited reduced expression of the anti-apoptotic intracellular protein Bcl-2 and significantly increased levels of apoptosis; the size and number of Peyer’s patches were also reduced, likely due to the significantly increased T-cell production of interferon (IFN)-γ. Moreover, the upregulated Th1 cytokine production may be in response to the intestinal enteric flora (Ohman et al. 2002, 2005). In addition, an increased frequency of CD4+Foxp3+ regulatory T cells (Tregs) was observed in Gαi2−/− mice, and this increased number of Tregs had no endogenous functional defect. However, the increased effective numbers of Tregs were unable to regulate the highly potent Gαi2−/− effector T cells in vitro and in vivo. Therefore, these cells cannot prevent the development of autoimmune diseases, such as colitis (Gotlind et al. 2011), because the Gαi2−/− T effector population comprises significantly higher number of cells with a CD4+CD62L−CD44+ effector memory phenotype than WT cells; these memory cells are more pro-inflammatory, more easily activated to proliferate and less susceptible to regulation by Tregs (Huang et al. 2003).
In addition to the Gαi/o and Gαq/11 family members, the Gα12/13 family of G proteins also has important regulatory roles in the T-cell life cycle (Coffield et al. 2004; Herroeder et al. 2009). Using Gα13 and Gα12 minigenes, the mutant could bind both Gα12 and Gα13 through the RGS domain and thereby prevent these receptors from transducing a signal to downstream effectors. Mice with T cells that exhibited genetically inactivated Gα12 and Gα13 have increased cell numbers, particularly CD4+ T cells in the lymph nodes, blood, and the thymus. In addition, these Gα12-Gα13-double-deficient CD4+ T cells showed an enhanced interaction with dendritic cells (DCs), which suggests that these G proteins have regulatory functions in CD4+ T-cell activation, proliferation and apoptosis (Herroeder et al. 2009).
G Proteins Are Involved in Leukocyte Migration
The coordinated retention and relocation of leukocytes have key roles in the development, maintenance and proper functioning of the immune system, and a loss in the control of leukocyte traffic might contribute to immune suppression and autoimmune diseases (Jin et al. 2008a; Moser et al. 2004). It is now well recognised that chemokines and their receptors are master controllers of leukocyte migration, and studies over the past 30 years also reveal that chemokine receptors represent a new subfamily of GPCRs. These chemokine receptors activate the Gαs, Gαi/o, Gαq/11 and Gα12/13 families of G proteins depending on the type and activation state of the respective cell (Bennett et al. 2011; Kunkel and Butcher 2002; Wettschureck and Offermanns 2005; Zlotnik et al. 2011).
The role of Gαi in chemotaxis has been extensively investigated, and studies using KO mice have revealed that Gαi has a crucial role in leukocyte migration (Chaffin and Perlmutter 1991; Jin et al. 2008b; Spangrude et al. 1985; Thompson et al. 2007). The irreversible blocking of Gαi-mediated signalling by pertussis toxin (PTX) strongly impairs lymphocyte migration in vitro (Spangrude et al. 1985) and causes the accumulation of mature T cells in the thymus and greatly reduced level of T cells in the peripheral lymphatic organs in vivo (Chaffin and Perlmutter 1991). The PTX-induced blocking of Gαi-mediated signalling also causes defective homing of the peripheral lymphocytes to the spleen, lymph nodes, and Peyer’s patches (Cyster and Goodnow 1995; Warnock et al. 1998). The results of a study that used Gαi2−/−- and Gαi3−/−-KO T cells indicate that Gαi2 is indispensable for T-cell migration and GTPγS incorporation upon CXCR3-stimulation and that the Gαi3- KO T cells display a significant increase in both GTPγS incorporation and migration when stimulated with CXCR3 agonists. More importantly, the increased GTPγS incorporation could be blocked by the Gαi3 protein in a dose-dependent manner (Thompson et al. 2007). Similarly, during the development of graft-versus-host disease, the deletion of Gαi2 hampered the trafficking of pathogenic T cells from the secondary lymphoid tissues to the inflammatory sites, and Gαi2−/− T cells displayed a defect in their response to CXCL10, CXCL11, and CCL5. In contrast, an aggravated rejection was induced in mice that were adoptively transferred with Gαi3-deficient T cells, and the absence of Gαi3 augmented the CXCL10- and CXCL11-induced chemotaxis of effector T cells and resulted in the homing preference of these cells to the liver and colon (Jin et al. 2008b). In neutrophils, Gαi2 is required for chemokine-induced arrest in response to CXCL1, and Gαi2−/− mice show significant defects in neutrophil recruitment in lipopolysaccharide (LPS)-induced inflammatory models (Zarbock et al. 2007). Although a deficiency in the Gαi2 protein does not affect neutrophil function, a significantly reduced recruitment of neutrophils into the microenvironment of the parasites in immunised Gαi2−/− mice is observed, and these signalling events are necessary for the recruitment of neutrophils that ultimately leads to the host-mediated killing of the larvae (Padigel et al. 2007). Furthermore, Gαi2 and not Gαi3 is essential for optimal CCL2- and C5a-induced recruitment of macrophages in acute inflammation (Wiege et al. 2012); however, not all effector functions of macrophages are mediated by Gαi2-specific signalling, and Gαi3 is able to act as a substitute for Gαi2 in C5aR-regulated phagocytosis and Gαi-dependent cytokine production (Fan et al. 2007). Interestingly, the Gα-interacting vesicle-associated protein (GIV), which is a recently discovered non-receptor guanine nucleotide exchange factor (GEF), can also trigger G protein activation (Garcia-Marcos et al. 2009). The Gαi3-GIV association is essential for macrophage chemotaxis through the enhancement of Akt activation and the remodelling of the actin cytoskeleton (Ghosh et al. 2008). Further studies are required to clarify the reciprocal function of Gαi2 and Gαi3 in leukocyte migration.
There is also evidence that the activation of Gαq/11-mediated signalling is involved in chemokine-induced leukocyte migration (Al-Aoukaty et al. 1998; Shi et al. 2007; Soede et al. 2001). Macrophage inflammatory protein-3α receptors can couple to Gαq/11 proteins to enhance a robust calcium response flux and induce the motility of interleukin (IL)-2-activated natural killer cells (Al-Aoukaty et al. 1998). In myeloid cells, Gαq/11 is required for integrin activation in the bone marrow and induces the integrin LFA-1-dependent aggregation of TAM2D2 T-cell hybridoma cells (Soede et al. 2001). More importantly, Gαq-deficient mice show defective calcium and chemotactic responses upon stimulation of neutrophils with N-formylmethionine leucyl-phenylalanine (fMLP) and CCL3 and upon stimulation of DCs with CCL2, CCL19, CCL21, and CXCL12. In contrast, the Gαq-null T-cell responses to CXCL12 and CCL19 are not affected (Shi et al. 2007). Thus, an alternative Gαq-dependent chemokine receptor pathway may control the migration of only a subset of leukocytes (Shi et al. 2007). The novel chemokine receptor signalling pathway appears to be critically important for the initiation of inflammatory responses because Gαq is required for the migration of DCs from the skin to the draining lymph nodes after fluorescein isothiocyanate sensitisation and for the emigration of monocytes from the bone marrow to the inflamed skin after contact sensitisation (Shi et al. 2007). In addition, the macrophages of Gα15- but not Gα11- and Gαq-deficient mice exhibit only a minor signalling defect in response to complement C5a (Davignon et al. 2000). Gα16, which is the human counterpart of Gα15, can also efficiently couple chemoattractant receptors to NF-κB activation, which suggests a potential function of Gα16 beyond the activation of PLCβ (Yang et al. 2001). However, contradictory results have been obtained for the roles of Gαq and Gαi2 in the regulation of T-cell migration. A study indicated that the knockdown of Gαq resulted in an approximately 80 % increase in CXCL12-induced T-cell migration, whereas the knockdown of Gαi2 inhibited CXCL12-induced T-cell migration (Ngai et al. 2008, 2009). Therefore, further studies are required to clarify the roles of Gαq and Gαi2 in the regulation of T-cell migration.
Due to the embryonic lethality of Gα12/Gα13-DKO, direct evidence for the involvement of the Gα12/13 subfamily in lymphocyte adhesion and migration is not yet available (Gu et al. 2002). However, the Gα12/13 effector RhoA has been repeatedly shown to be involved in lymphocyte traffic. The murine Rho-specific GEF Lsc can couple Gα13 to activate RhoA. The B cells of Lsc-deficient mice exhibit impaired actin polymerisation and motility and abnormal homing (Girkontaite et al. 2001). In addition, defective migration is also observed in mice that lack the orphan G protein-coupled receptor G2A, which may be directly coupled to Gα13. G2A-deficient macrophages and T cells exhibit reduced migration towards lysophosphatidylcholine (LPC), whereas G2A overexpression in a macrophage cell line enhances the migration of these cells towards LPC (Murakami et al. 2004; Radu et al. 2004). T- or B-cell-specific Gα12/Gα13-DKO mice were generated using the Cre-loxP system. The resultant Gα12/Gα13-DKO MZB cells exhibit significantly increased migration towards sphingosine 1-phosphate, calf serum, and mouse serum, whereas the serum-induced migration was not altered in the follicular B cells (Rieken et al. 2006). Similarly, Gα12/Gα13-DKO CD4+ T cells also exhibit increased adhesiveness and enhanced lymph node entry (Herroeder et al. 2009).
Neutrophil polarisation and directed migration can also be mediated by the G protein βγ-subunits (Neptune et al. 1999). The complement C5a-induced migration of J774A.1 mouse macrophages critically depends on Gβ2, but not on Gβ1, Gαi2, or Gαi3 (Hwang et al. 2004). PI3 Kγ, PLCβ2 and PLCβ3 are intracellular effectors of the dissociated Gβγ-subunits in neutrophils. Although neutrophils from mice lacking PLCβ2 or PLCβ3 exhibit normal and even enhanced chemotactic responses, the migration of PI3 Kγ-deficient neutrophils is severely impaired, and these cells fail to accumulate at the sites of inflammation in a septic peritonitis model (Li et al. 2000).
Cytokine Production Is Regulated by G Proteins
In general, the physiologic function of all immune responses is to eliminate microbes and other foreign antigens. It has recently become obvious that GPCRs and heterotrimeric G proteins can trigger different aspects of innate and adaptive immunity and exert important modulatory effects on the immune cell effector functions, including cytokine production, lymphocyte differentiation, phagocytosis, and mediator release (Wettschureck and Offermanns 2005).
The treatment of normal mice with PTX inhibited Gαi protein signalling and thus mimics the conditions that are observed in Gαi2-deficient mice (Ryan et al. 1998). Various effects of PTX on the immune system have been reported, including a polarised Th1-type immune response, an enhanced capacity of splenocytes to produce IL-12, tumour necrosis factor (TNF)-α, IFN-γ and IL-2 in response to both microbial and non-microbial stimuli, and an augmented expression of co-stimulatory molecules, such as B7-1 and B7-2 on macrophages and their counter-receptor CD28 on T cells (Shive et al. 2000). These findings are further confirmed by the fact that untreated Gαi2−/− mice also exhibit enhanced production of IL-12 and TNF-α by splenocytes and of IL-12 p40 by purified splenic CD8α+ lymphoid DCs (He et al. 2000). Moreover, T cells from Gαi2−/− but not Gαi3−/− mice exhibit hyperresponsiveness in their Th1-type cytokine production after their activation through the TCR, and Gαi2−/− T cells have a relaxed co-stimulatory requirement for IL-2 secretion and proliferation compared to WT cells (Huang et al. 2003). In addition to the induction of Th1 responses, recent studies found that the loss of Gαi2 can also promote a Th17 phenotype with significantly greater levels of IL-17, whereas CD11c+ bone marrow-derived DCs produce higher levels of the inflammatory cytokine IL-23 and a minimal IL-10 response to CpG (Pena et al. 2009). More importantly, the B-cell population of Gαi2−/− mice is functionally deficient in LPS-induced proliferation and IL-10 production, which indicates that Gαi2 is required for the development of IL-10-producing B cells (the potential regulatory B cells) (Dalwadi et al. 2003). These immune function abnormalities in Gαi2−/− mice might contribute to lymphocyte-mediated autoimmunity in these mice.
The requirement of Gαq/11-mediated signalling for T-cell activation was first demonstrated in RGS2-deficient mice. In addition to a proliferative defect, the RGS2−/− T cells produced significantly lower levels of the T-cell growth factor IL-2 and induced an impaired antiviral immunity in vivo (Oliveira-Dos-Santos et al. 2000). The activation of the murine thromboxane A2 receptor, which is typically coupled to the Gαq/11 family of G proteins, impairs the DC-T cell adhesion and inhibits the DC-dependent proliferation of T cells (Kabashima et al. 2003). The abovementioned indirect evidence indicates that Gαq/11 might be a negative regulator of acquired immunity. Recent studies with KO mice further support the involvement of Gαq/11 in immune cell effector functions (Bueno et al. 2006; Misra et al. 2010; Ngai et al. 2008). Bacterial superantigens can bypass Lck-dependent TCR signalling through the activation of a Gα11-dependent PLCβ-mediated pathway. This alternative signalling pathway leads to the activation of Erk and protein kinase C and to the influx of Ca2+ and induces a substantial secretion of IL-2 (Bueno et al. 2006). Similarly, the siRNA targeting of Gαq demonstrates a specific role of Gαq in TCR signalling. Gαq-deficient Jurkat TAg T cells display a reduced activation of Lck but paradoxically show sustained Erk1/2 phosphorylation. Consistent with this finding, Gαq−/− primary T cells also show reduced proximal LAT phosphorylation, sustained Erk1/2 phosphorylation and augmented immune responses, including increased secretion of IL-2, IL-5, IL-12 and TNF-α (Ngai et al. 2008). Furthermore, upon anti-IgM stimulation, the levels of phospho-Akt, phospho-PLCγ2 and phospho-Erk are significantly increased in Gαq−/− B cells, which suggests that there is an increased activation of BCR-mediated signalling in Gαq−/− B cells (Misra et al. 2010). In contrast, mutant Gα16 transfectants display an inhibition of TCR/CD3-mediated signalling and deficiencies in IL-2 and IL-10 production and CD69 expression (Zhou et al. 1998).
In contrast, the inactivation of Gαs-mediated signalling exhibits an immunosuppressive effect. CD4+ T cells in Gαs-conditional-KO mice exhibit decreased production of cAMP, reduced Ca2+ influx, lower secretion of IL-17, IL-22 and IFN-γ, and normal IL-4 production and cannot mount an antigen-specific Th17 response upon oral CT/OVA immunisation. Due to the selective modulation of Th17 and Th1 differentiation, the adoptive transfer of naive Gαs−/− CD4+ T cells into RAG1−/− recipients provokes minimal colonic inflammation, and the mice ultimately fail to develop colitis (Li et al. 2012). The two members of the Gα12 family, Gα12 and Gα13, were ubiquitously expressed and have been reported to mediate actin polymerisation during T-cell activation (Offermanns et al. 1997). The activation of RhoA by Gα12 and Gα13 is mediated by a subgroup of GEFs for Rho, and both RhoGEF and RhoA have been shown to affect T-cell function (Galandrini et al. 1997; Girkontaite et al. 2001). The indirect evidence suggests that Gα12-mediated signalling might play an important regulatory role in T-cell activation.
G Proteins and Autoimmune Diseases
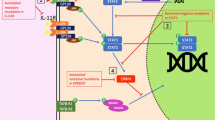
Gαi2-deficient mice exhibit a pro-inflammatory phenotype and can develop severe immunological problems, such as fatal inflammatory bowel disease (IBD) (Rudolph et al. 1995). IBD is a collective term that refers to chronic, autoimmune, and inflammatory diseases of the bowel, mainly ulcerative colitis and Crohn’s disease (Baumgart and Carding 2007). The Gαi2−/− mouse is a well-established model for colitis and is characterised by a Th1 CD4+ T-cell response with an increased production of IFN-γ and increased levels of IL-1, IL-6 and TNF-α in the inflamed tissue (Hornquist et al. 1997; Ohman et al. 2002). The absence of Gαi2 also results in defective arachidonate release and prostaglandin E2 production in intestinal and colonic subepithelial myofibroblasts and dysregulation of the epithelial cell barrier (Edwards and Smock 2006; Edwards et al. 2008; Saha et al. 1998). In addition, the loss of Gαi2 results in elevated IL-12 levels and insufficient IL-10 secretion by macrophages and splenic CD8α+ DCs (He et al. 2000; Pena et al. 2009). Furthermore, the null mutation of Gαi2 can cause a reduction in the MZB and peritoneal B-1b cell subpopulations (Dalwadi et al. 2003) and Treg cell non-function (Gotlind et al. 2011). These facts indicate the multiple requirements for Gαi2 in mucosal immune responses. Gαi2-deficient mice can spontaneously develop colitis by the age of 16–21 weeks, and 30–40 % of the animals develop nonpolypoid adenocarcinoma, which is a typical character of human ulcerative colitis (Rudolph et al. 1995). Genetic differences between mouse strains might affect the susceptibility of Gαi2−/− mice to colitis: Gαi2−/− C57BL/6 mice were relatively resistant to colitis, whereas Gαi2−/− 129/Sv mice developed IBD earlier and with greater frequency and severity (Bjursten et al. 2004). Genetic linkage studies in humans have mapped the Gαi2 gene within an IBD-susceptible locus at chromosome 3p21, which raises the possibility that Gαi2 is one of the candidate genes that contribute to IBD development in humans (Fig. 1) (Hampe et al. 2001). Remarkably, genetically engineered mice raised in germ-free conditions on a 129/Sv strain background appear to be less susceptible to IBD-like diseases compared with mice housed in SPF conditions, and germ-free studies have also revealed the key role of the microbiota in the development of colitis (Arthur and Jobin 2011; Taurog et al. 1994). These findings have led to an intense interest in the interactions between the intrinsic (genetic) host defects and extrinsic (microbiota) factors. One study has shown that an ecological disorder of the bacterial community, which is also named “dysbiotic microbiota”, can cause IBD and that the symptoms are more severe in genetic animals models (Neish 2009). Consistently, an antibiotic treatment can also induce remission in IBD patients (Khan et al. 2011). Furthermore, the autoantibodies detected in IBD models are very likely the result of cross-reactivity with bacterial antigens (Targan and Karp 2005).

Possible immune pathogenesis of Gαi2−/− colitis. The loss of Gαi2 can lead to an amplification of Th17/Th1 responses, which are likely mediated by a defect in the epithelium tight junctions. IL-17 is fed back to the macrophages, which then upregulate CXCL1 secretion, and induces neutrophil ingress. The lack of appropriate regulation by Tregs further exacerbates the Gαi2−/− colitis
A previous study with Gαq−/− chimeric mice, which were generated through the reconstitution of lethally irradiated C57BL/6 J recipient mice with Gαq−/− bone marrow, demonstrated that Gαq−/− chimeras could spontaneously develop systemic autoimmunity with multi-organ involvement, including the production of autoreactive antibodies by Gαq−/− B cells, the deposition of IgG2a- and IgG2c-containing immune complexes in the kidney, thrombotic microangiopathy, the reduction in the numbers of red blood cells, synovitis, bone resorption, exostotic bone development, and osteolytic activity (Misra et al. 2010). These pathological features may be manifestations of diseases in Lupus or arthritis patients (Misra et al. 2010). Because there is over 99 % identity between the human and mouse Gnaq genes, Gαq might also play a role in the pathogenesis of human rheumatoid arthritis (RA). We recently showed that the expression of Gαq in the PBLs from RA patients is significantly decreased and that the expression level of Gαq mRNA in PBLs from RA patients strongly correlates with RA disease activity (DAS28), anti-cyclic citrullinated protein antibodies, C-reactive protein and rheumatoid factor (Elgbratt et al. 2012). Our results support the hypothesis that Gαq may be involved in the pathogenesis and progression of RA through the regulation of the apoptosis and survival of PBLs (Fig. 2).

Possible immune pathogenesis of Gαq−/− arthritis. Gαq has a critical intrinsic role in the maintenance of the immunological tolerance of peripheral lymphocytes. Gαq modulates the peripheral B-cell development and appears to control the numbers of T1-derived MZB cell precursors and mature MZB cells. Gαq−/− T and B cells are more likely to survive than WT lymphocytes and exhibit significant proliferation. These abnormal Gαq−/− lymphocytes might contribute to the development of arthritis
The data obtained using G2A−/− mice present indirect evidence that supports the hypothesis that the inactivation of Gα13 may be involved in the development of autoimmune-like syndrome. Older G2A−/− mice present multiple signs of autoimmunity with heavy lymphocytic infiltration in various organs, increased immunoglobulin levels, the deposition of immunoglobulin complexes in the glomeruli and the presence of anti-nuclear autoantibodies. Together with the emergence of lymphadenopathy in ageing G2A−/− mice, these observations suggest the development of a late-onset non-organ-specific autoimmune syndrome (Hampe et al. 2001). Similarly, T-cell-specific Gα12/Gα13-DKO mice also exhibit increased CD4+ T-cell proliferation and infiltration, enhanced proinflammatory cytokine production and the deposition of IgG-containing immune complexes in the kidney (Herroeder et al. 2009). These facts indicate that Gα13−/− mice may be associated with increased susceptibility and severity of T-cell-mediated abnormal immune responses and autoimmune disease (Hampe et al. 2001; Herroeder et al. 2009).
Concluding Remarks
The past several years have seen an explosion of data that uncover the roles of heterotrimeric G proteins signalling in the regulation of immune cell function and in the development of diverse autoimmune-like syndromes. Many of these insights were derived from studies that used gene-targeted mice. An important point has to be taken into consideration: chemokines are a superfamily that contains 48 ligands that bind to 19 different GPCRs. In addition to their best recognised function, which is the induction of immune cell migration, chemokines have many functions, including the regulation of immune cell survival and effector responses. With the availability of KO mice, the significance of many aspects of G protein-mediated signalling in the innate and adaptive immune response has been studied, including the roles of G proteins in autoimmunity. It is worth paying more attention to the function of various G proteins in the development and progression of human autoimmune diseases by studying the relationship between G proteins and human autoimmune diseases and verifying the internal links epidemiologically. Due to the functional diversity of G proteins in the immune system, the study of the crosstalk between multiple subforms of G proteins-mediated signalling, such as Gαq and Gα11, will likely lead to new insights that will help elucidate their function in the maintenance of immune homeostasis. In conclusion, the expression of G proteins in immune cells, such as granulocytes and lymphocytes, represents an important regulatory component of the intracellular signalling pathways that are induced by GPCRs. Thus, the molecular elucidation of the functions of G proteins in immunity might pave the way for the development of novel therapeutics for the treatment of autoimmune diseases, and the modulation of G protein signalling downstream of the receptor might lead to the development of novel drugs with better efficacy and/or fewer side effects.
References
Al-Aoukaty A, Rolstad B, Giaid A et al (1998) MIP-3alpha, MIP-3beta and fractalkine induce the locomotion and the mobilization of intracellular calcium, and activate the heterotrimeric G proteins in human natural killer cells. Immunology 95:618–624
Arthur JC, Jobin C (2011) The struggle within: microbial influences on colorectal cancer. Inflamm Bowel Dis 17:396–409
Baumgart DC, Carding SR (2007) Inflammatory bowel disease: cause and immunobiology. Lancet 369:1627–1640
Bennett LD, Fox JM, Signoret N (2011) Mechanisms regulating chemokine receptor activity. Immunology 134:246–256
Bjursten M, Hultgren OH, Hultgren Hornquist E (2004) Enhanced pro-inflammatory cytokine production in Galphai2-deficient mice on colitis prone and colitis resistant 129 Sv genetic backgrounds. Cell Immunol 228:77–80
Bockaert J, Claeysen S, Becamel C et al (2002) G protein-coupled receptors: dominant players in cell–cell communication. Int Rev Cytol 212:63–132
Browne CD, Del Nagro CJ, Cato MH et al (2009) Suppression of phosphatidylinositol 3,4,5-trisphosphate production is a key determinant of B cell anergy. Immunity 31:749–760
Bueno C, Lemke CD, Criado G et al (2006) Bacterial superantigens bypass Lck-dependent T cell receptor signaling by activating a Galpha11-dependent, PLC-beta-mediated pathway. Immunity 25:67–78
Cantrell D (2002) Protein kinase B (Akt) regulation and function in T lymphocytes. Semin Immunol 14:19–26
Chaffin KE, Perlmutter RM (1991) A pertussis toxin-sensitive process controls thymocyte emigration. Eur J Immunol 21:2565–2573
Coffield VM, Helms WS, Jiang Q et al (2004) Galpha13 mediates a signal that is essential for proliferation and survival of thymocyte progenitors. J Exp Med 200:1315–1324
Cyster JG, Goodnow CC (1995) Pertussis toxin inhibits migration of B and T lymphocytes into splenic white pulp cords. J Exp Med 182:581–586
Dalwadi H, Wei B, Schrage M et al (2003) B cell developmental requirement for the G alpha i2 gene. J Immunol 170:1707–1715
Davignon I, Catalina MD, Smith D et al (2000) Normal hematopoiesis and inflammatory responses despite discrete signaling defects in Galpha15 knockout mice. Mol Cell Biol 20:797–804
Druey KM (2009) Regulation of G-protein-coupled signaling pathways in allergic inflammation. Immunol Res 43:62–76
Edwards RA, Smock AZ (2006) Defective arachidonate release and PGE2 production in Gi alpha2-deficient intestinal and colonic subepithelial myofibroblasts. Inflamm Bowel Dis 12:153–165
Edwards RA, Wang K, Davis JS et al (2008) Role for epithelial dysregulation in early-onset colitis-associated colon cancer in Gi2-alpha−/− mice. Inflamm Bowel Dis 14:898–907
Elgbratt K, Bjursten M, Willen R et al (2007) Aberrant T-cell ontogeny and defective thymocyte and colonic T-cell chemotactic migration in colitis-prone Galphai2-deficient mice. Immunology 122:199–209
Elgbratt K, Jansson A, Hultgren-Hornquist E (2012) A quantitative study of the mechanisms behind thymic atrophy in Galphai2-deficient mice during colitis development. PLoS ONE 7:e36726
Entschladen F, Zanker KS, Powe DG (2011) Heterotrimeric G protein signaling in cancer cells with regard to metastasis formation. Cell Cycle 10:1086–1091
Fan H, Williams DL, Zingarelli B et al (2007) Differential regulation of lipopolysaccharide and Gram-positive bacteria induced cytokine and chemokine production in macrophages by Galpha(i) proteins. Immunology 122:116–123
Galandrini R, Henning SW, Cantrell DA (1997) Different functions of the GTPase Rho in prothymocytes and late pre-T cells. Immunity 7:163–174
Garcia-Marcos M, Ghosh P et al (2009) GIV is a nonreceptor GEF for G alpha i with a unique motif that regulates Akt signaling. Proc Natl Acad Sci USA 106:3178–3183
Ghosh P, Garcia-Marcos M, Bornheimer SJ et al (2008) Activation of Galphai3 triggers cell migration via regulation of GIV. J Cell Biol 182:381–393
Girkontaite I, Missy K, Sakk V et al (2001) Lsc is required for marginal zone B cells, regulation of lymphocyte motility and immune responses. Nat Immunol 2:855–862
Goldsmith ZG, Dhanasekaran DN (2007) G protein regulation of MAPK networks. Oncogene 26:3122–3142
Gotlind YY, Raghavan S, Bland PW et al (2011) CD4 + FoxP3 + regulatory T cells from Galphai2−/− mice are functionally active in vitro, but do not prevent colitis. PLoS ONE 6:e25073
Gu JL, Muller S, Mancino V et al (2002) Interaction of G alpha(12) with G alpha(13) and G alpha(q) signaling pathways. Proc Natl Acad Sci USA 99:9352–9357
Hampe J, Lynch NJ, Daniels S et al (2001) Fine mapping of the chromosome 3p susceptibility locus in inflammatory bowel disease. Gut 48:191–197
He J, Gurunathan S, Iwasaki A et al (2000) Primary role for Gi protein signaling in the regulation of interleukin 12 production and the induction of T helper cell type 1 responses. J Exp Med 191:1605–1610
Herroeder S, Reichardt P, Sassmann A et al (2009) Guanine nucleotide-binding proteins of the G12 family shape immune functions by controlling CD4 + T cell adhesiveness and motility. Immunity 30:708–720
Hildeman D, Jorgensen T, Kappler J et al (2007) Apoptosis and the homeostatic control of immune responses. Curr Opin Immunol 19:516–521
Holtzman MJ, Green JM, Jayaraman S et al (2000) Regulation of T cell apoptosis. Apoptosis 5:459–471
Hornquist CE, Lu X, Rogers-Fani PM et al (1997) G(alpha)i2-deficient mice with colitis exhibit a local increase in memory CD4 + T cells and proinflammatory Th1-type cytokines. J Immunol 158:1068–1077
Huang TT, Zong Y, Dalwadi H et al (2003) TCR-mediated hyper-responsiveness of autoimmune Galphai2(−/−) mice is an intrinsic naive CD4(+) T cell disorder selective for the Galphai2 subunit. Int Immunol 15:1359–1367
Hwang JI, Fraser ID, Choi S et al (2004) Analysis of C5a-mediated chemotaxis by lentiviral delivery of small interfering RNA. Proc Natl Acad Sci USA 101:488–493
Jiang M, Boulay G, Spicher K et al (1997) Inactivation of the G alpha i2 and G alpha o genes by homologous recombination. Receptors Channels 5:187–192
Jin Y, Wu MX (2008) Requirement of Galphai in thymic homing and early T cell development. Mol Immunol 45:3401–3410
Jin T, Xu X, Hereld D (2008a) Chemotaxis, chemokine receptors and human disease. Cytokine 44:1–8
Jin YZ, Thompson BD, Zhou ZY et al (2008b) Reciprocal function of Gαi2 and Gαi3 in graft-versus-host disease. Eur J Immunol 38:1988–1998
Kabashima K, Murata T, Tanaka H et al (2003) Thromboxane A2 modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat Immunol 4:694–701
Khan KJ, Ullman TA, Ford AC et al (2011) Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol 106:661–673
Koelle MR (2006) Heterotrimeric G protein signaling: getting inside the cell. Cell 126:25–27
Kunkel EJ, Butcher EC (2002) Chemokines and the tissue-specific migration of lymphocytes. Immunity 16:1–4
Li Z, Jiang H, Xie W et al (2000) Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science 287:1046–1049
Li X, Murray F, Koide N et al (2012) Divergent requirement for Galphas and cAMP in the differentiation and inflammatory profile of distinct mouse Th subsets. J Clin Invest 122:963–973
Meyer-Bahlburg A, Andrews SF, Yu KO et al (2008) Characterization of a late transitional B cell population highly sensitive to BAFF-mediated homeostatic proliferation. J Exp Med 205:155–168
Misra RS, Shi G, Moreno-Garcia ME et al (2010) G alpha q-containing G proteins regulate B cell selection and survival and are required to prevent B cell-dependent autoimmunity. J Exp Med 207:1775–1789
Molon B, Gri G, Bettella M et al (2005) T cell costimulation by chemokine receptors. Nat Immunol 6:465–471
Moser B, Wolf M, Walz A et al (2004) Chemokines: multiple levels of leukocyte migration control. Trends Immunol 25:75–84
Murakami N, Yokomizo T, Okuno T et al (2004) G2A is a proton-sensing G-protein-coupled receptor antagonized by lysophosphatidylcholine. J Biol Chem 279:42484–42491
Neish AS (2009) Microbes in gastrointestinal health and disease. Gastroenterology 136:6–80
Neptune ER, Iiri T, Bourne HR (1999) Galphai is not required for chemotaxis mediated by Gi-coupled receptors. J Biol Chem 274:2824–2828
Ngai J, Methi T, Andressen KW et al (2008) The heterotrimeric G-protein alpha-subunit Galphaq regulates TCR-mediated immune responses through an Lck-dependent pathway. Eur J Immunol 38:3208–3218
Ngai J, Inngjerdingen M, Berge T et al (2009) Interplay between the heterotrimeric G-protein subunits Galphaq and Galphai2 sets the threshold for chemotaxis and TCR activation. BMC Immunol 10:27
Offermanns S, Mancino V, Revel JP et al (1997) Vascular system defects and impaired cell chemokinesis as a result of Galpha13 deficiency. Science 275:533–536
Ohman L, Franzen L, Rudolph U et al (2002) Regression of Peyer’s patches in G alpha i2 deficient mice prior to colitis is associated with reduced expression of Bcl-2 and increased apoptosis. Gut 51:392–397
Ohman L, Astrom RG, Hultgren Hornquist E (2005) Impaired B cell responses to orally administered antigens in lamina propria but not Peyer’s patches of Galphai2-deficient mice prior to colitis. Immunology 115:271–278
Oliveira-Dos-Santos AJ, Matsumoto G, Snow BE et al (2000) Regulation of T cell activation, anxiety, and male aggression by RGS2. Proc Natl Acad Sci USA 97:12272–12277
Padigel UM, Stein L, Redding K et al (2007) Signaling through Galphai2 protein is required for recruitment of neutrophils for antibody-mediated elimination of larval Strongyloides stercoralis in mice. J Leuk Biol 81:1120–1126
Pena JA, Thompson-Snipes L, Calkins PR et al (2009) Alterations in myeloid dendritic cell innate immune responses in the Galphai2-deficient mouse model of colitis. Inflamm Bowel Dis 15:248–260
Radu CG, Yang LV, Riedinger M et al (2004) T cell chemotaxis to lysophosphatidylcholine through the G2A receptor. Proc Natl Acad Sci USA 101:245–250
Rieken S, Sassmann A, Herroeder S et al (2006) G12/G13 family G proteins regulate marginal zone B cell maturation, migration, and polarization. J Immunol 177:2985–2993
Rudolph U, Finegold MJ, Rich SS et al (1995) Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat Genet 10:143–150
Ryan M, McCarthy L, Rappuoli R et al (1998) Pertussis toxin potentiates Th1 and Th2 responses to co-injected antigen: adjuvant action is associated with enhanced regulatory cytokine production and expression of the co-stimulatory molecules B7–1, B7–2 and CD28. Int Immunol 10:651–662
Saha C, Nigam SK, Denker BM (1998) Involvement of Galphai2 in the maintenance and biogenesis of epithelial cell tight junctions. J Biol Chem 273:21629–21633
Shi G, Partida-Sánchez S, Misra RS et al (2007) Identification of an alternative Gαq-dependent chemokine receptor signal transduction pathway in dendritic cells and granulocytes. J Exp Med 204:2705–2718
Shive CL, Hofstetter H, Arredondo L et al (2000) The enhanced antigen-specific production of cytokines induced by pertussis toxin is due to clonal expansion of T cells and not to altered effector functions of long-term memory cells. Eur J Immunol 30:2422–2431
Soede RD, Zeelenberg IS, Wijnands YM et al (2001) Stromal cell-derived factor-1-induced LFA-1 activation during in vivo migration of T cell hybridoma cells requires Gq/11, RhoA, and myosin, as well as Gi and Cdc42. J Immunol 166:4293–4301
Spangrude GJ, Sacchi F, Hill HR et al (1985) Inhibition of lymphocyte and neutrophil chemotaxis by pertussis toxin. J Immunol 135:4135–4143
Strasser A, Bouillet P (2003) The control of apoptosis in lymphocyte selection. Immunol Rev 193:82–92
Targan SR, Karp LC (2005) Defects in mucosal immunity leading to ulcerative colitis. Immunol Rev 206:296–305
Taurog JD, Richardson JA, Croft JT et al (1994) The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med 180:2359–2364
Thien M, Phan TG, Gardam S et al (2004) Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity 20:785–798
Thompson BD, Jin Y, Wu KH et al (2007) Inhibition of G alpha i2 activation by G alpha i3 in CXCR3-mediated signaling. J Biol Chem 282:9547–9555
von Boehmer H, Melchers F (2010) Checkpoints in lymphocyte development and autoimmune disease. Nat Immunol 11:14–20
Wang Y, Li Y, He Y et al (2011) Expression of G protein alphaq Subunit is decreased in lymphocytes from rheumatoid arthritis patients and is correlated with disease activity. Scand J Immunol. doi:10.1111/j.1365-3083.2011.02635.x
Warnock RA, Askari S, Butcher EC et al (1998) Molecular mechanisms of lymphocyte homing to peripheral lymph nodes. J Exp Med 187:205–216
Wettschureck N, Offermanns S (2005) Mammalian G proteins and their cell type specific functions. Physiol Rev 85:1159–1204
Wiege K, Le DD, Syed SN et al (2012) Defective macrophage migration in Galphai2- but not Galphai3-deficient mice. J Immunol 189:980–987
Xu G, Shi Y (2007) Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res 17:759–771
Yanamadala V, Negoro H, Denker BM (2009) Heterotrimeric G proteins and apoptosis: intersecting signaling pathways leading to context dependent phenotypes. Curr Mol Med 9:527–545
Yang M, Sang H, Rahman A et al (2001) G alpha 16 couples chemoattractant receptors to NF-kappa B activation. J Immunol 166:6885–6892
Zarbock A, Deem TL, Burcin TL et al (2007) Galphai2 is required for chemokine-induced neutrophil arrest. Blood 110:3773–3779
Zhang Y, Finegold MJ, Jin Y et al (2005) Accelerated transition from the double-positive to single-positive thymocytes in G alpha i2-deficient mice. Int Immunol 17:233–243
Zhou J, Stanners J, Kabouridis P et al (1998) Inhibition of TCR/CD3-mediated signaling by a mutant of the hematopoietically expressed G16 GTP-binding protein. Eur J Immunol 28:1645–1655
Zlotnik A, Burkhardt AM, Homey B (2011) Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol 11:597–606
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC) grants 30830094 and 30972678 to Dr Guixiu Shi.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Wang, Y., Li, Y. & Shi, G. The Regulating Function of Heterotrimeric G Proteins in the Immune System. Arch. Immunol. Ther. Exp. 61, 309–319 (2013). https://doi.org/10.1007/s00005-013-0230-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-013-0230-5