
Summary
A new PK variant with moderate hemolytic anemia is described. The enzymes of the nonanemic parents show sigmoidal reaction kinetics, with normal kinetic parameters, but differ with respect to nucleotide specificity, thermostability, and the concentrations of the glycolytic intermediates in the erythrocytes.
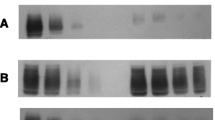
The most characteristic features of the patient's (daughter) enzyme are a 30% activity, hyperbolic reaction kinetics and only two bands in the SDS-gel electrophoresis instead of three bands observed with the parental enzymes. Moreover, the pH-optimum is shifted to the acidic range, the affinity for PEP and ADP is decreased, ATP inhibition is negligible and FDP-activation is roughly ten times smaller than with controls. The concentrations of 2.3-DPG, 2-PG and PEP in the erythrocyte are increased, but ATP decreased.
As there is no consanguinity in the parents and their enzymes are different this PK mutant can be considered to be compound-heterozygous for two different mutant PK alleles.
Similar content being viewed by others


References
Beutler E (1975) Red cell metabolism: A manual of biochemical methods, 2nd ed. Grune & Stratton, New York
Dacie JV, Lewis SM (1975) Practical haematology, 5th ed. Churchill Livingstone, Edinburgh
Ericson H, Verdier CH de (1972) Modified method for the determination of 2.3-diphosphoglycerate in erythrocytes. Scand J Clin Lab Invest 29: 85–90
International Committee for Standardization in Haematology (1979) Recommended methods for the characterization of red cell pyruvate kinase variants. Br J Haematol 43: 275–286
Ishida Y, Miwa S, Fujii H, Fujinami N, Fakegawa S, Yamoto D, (1981) Thirteen cases of pyruvate kinase deficiency found in Japan. Am J. Hemat 10:239–250
Kahn A, Marie J, Garreau H, Sprengers ED (1978) The genetic system of the L-type pyruvate kinase forms in man. Subunit structure, interrelation and kinetic characteristics of the pyruvate kinase enzymes from erythrocytes and liver. Biochim Biophys Acta 523: 59–74
Lämmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685
Lakomek M, Schröter W, Winkler H (1980) Red cell pyruvate kinase deficiency: An optimised assay. Clin Chim Acta 108: 31–40
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193: 265–275
Marie J, Kahn A, Boivin P (1977) Human erythrocyte pyruvate kinase. Total purification and evidence for its antigenic identity with L-type enzyme. Biochim Biophys Acta 481: 96–104
Mentzer WC (1981) Pyruvate kinase deficiency and disorders of glycolysis. In: Hematology of infancy and childhood. WB Saunders Eastbourne, pp 566–607
Minakami S, Suzuki C, Saito T, Yoshikawa H (1965) Studies on erythrocyte glycolysis. I. Determination of the glycolytic intermediates in human erythrocytes. J Biochem (Tokyo) 58: 543–550
Schröter W, Lakomek M, Scharnetzky M, Tillmann W, Winkler H (1982) Pyruvate kinase “Göttingen1,2”: Congenital hemolytic anemia, evidence of double heterozygosity, and lack of enzyme cooperativity. Hum Genet 60: 381–386
Staal GEJ, Rijksen G, Vlug AMC, Vromen-van den Bos B, Akkerman JWN, Gorter G, Dierick J, Petermans M (1982) Extreme deficiency of L-type pyruvate kinase with moderate clinical expression. Clin Chim Acta 118: 241–253
Valentine WN, Tanaka KR, Miwa S (1961) A specific erythrocyte glycolytic enzyme defect (pyruvate kinase) in three subjects with congenital non-spherocytic hemolytic anemia. Trans Assoc Am Physicians 74: 100–110
Zanella A, Rebulla P, Vullo C, Izzo C, Tedesco F, Sircha G (1978) Hereditary pyruvate kinase deficiency: Role of the abnormal enzyme in red cell pathophysiology. Br J Haematol 40: 551–562
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Lakomek, M., Winkler, H., Scharnetzky, M. et al. Erythrocyte pyruvate kinase deficiency: Characterization of a new variant (PK “Aarau”). Blut 48, 123–129 (1984). https://doi.org/10.1007/BF00320334
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF00320334