
Abstract
Purpose of review
Pharmacological treatment options for hypertrophic cardiomyopathy (HCM) are currently limited and comprise non-disease specific therapies such as β-blockers, non-dihydropyridine calcium channel blockers, and disopyramide. These agents that offer a variable degree of symptomatic relief, often suboptimal, are often limited by side-effects and fail to address the key molecular abnormalities of the disease.
Recent findings
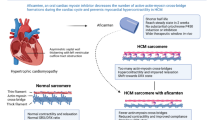
Mavacamten is a novel, first-in-class, allosteric inhibitor of cardiac myosin ATPase, which reduces actin-myosin cross-bridge formation, thereby reducing myocardial contractility and improving myocardial energetic consumption in experimental HCM models. Following a successful Phase 2 study, the recently published phase III, placebo-controlled, randomized EXPLORER-HCM trial demonstrated the efficacy and safety of mavacamten in reducing left ventricular outflow tract obstruction and ameliorating exercise capacity, New York Heart Association functional class and health status in patients with obstructive HCM.
Summary
Mavacamten represents the first agent specifically developed for HCM successfully tested in a Phase III trial, to be registered soon for clinical use, representing a radical change of paradigm in the pharmacological treatment of HCM.

Similar content being viewed by others


References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation. 1995;92:785–9.
Olivotto I, Girolami F, Nistri S, Rossi A, Rega L, Garbini F, et al. The many faces of hypertrophic cardiomyopathy: from developmental biology to clinical practice. J Cardiovasc Transl Res. 2009;2:349–67.
Doi YL, Deanfield JE, McKenna WJ, Dargie HJ, Oakley CM, Goodwin JF. Echocardiographic differentiation of hypertensive heart disease and hypertrophic cardiomyopathy. Br Heart J. 1980;44:395–400.
Pearson AC, Pasierski TJ, Orsinelli DA. Systolic anterior motion of the mitral chordae tendineae: prevalence and clinical and Doppler-echocardiographic features. Am Heart J. 1996;131:748–53.
Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Circulation. 1995;92:785–9.
Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC Guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2020;142:e558–631.
Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. European Society of Cardiology Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J. 2014;35:2733–79.
Maron MS, Olivotto I, Betocchi S, Casey SA, Lesser JR, Losi MA, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. Rev Port Cardiol. 2003;348:295–303.
Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation. 2018;138(14):1387–98.
Ammirati E, Contri R, Coppini R, Cecchi F, Frigerio M, Olivotto I. Pharmacological treatment of hypertrophic cardiomyopathy: current practice and novel perspectives. Eur J Heart Fail. 2016;18:1106–18.
Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AFL, Michels M, Ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail. 2015;3(11):896–905.
Wells S, Rowin EJ, Boll G, Rastegar H, Wang W, Maron MS, et al. Clinical profile of nonresponders to surgical myectomy with obstructive hypertrophic cardiomyopathy. Am J Med. 2018;131(6):e235–9.
Kim LK, Swaminathan RV, Looser P, Minutello RM, Wong SC, Bergman G, et al. Hospital volume outcomes after septal myectomy and alcohol septal ablation for treatment of obstructive hypertrophic cardiomyopathy: US nationwide inpatient database, 2003-2011. JAMA Cardiol. 2016;1(3):324–32.
Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, et al. Research priorities in hypertrophic cardiomyopathy: report of a working group of the National Heart, lung, and blood institute. Circulation. 2010;122(11):1130–3.
Elliott PM. Evolving story of clinical trials in hypertrophic cardiomyopathy. Circ Heart Fail. 2018;11(1):e004572.
Spudich JA. The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem Soc Trans. 2015;43(1):64–72.
Trivedi DV, Adhikari AS, Sarkar SS, Ruppel KM, Spudich JA. Hypertrophic cardiomyopathy and the myosin mesa: viewing an old disease in a new light. Biophys Rev. 2018;10(1):27–48.
• Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. Heart disease: a small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351(6273):617–21 Findings from this study suggest that MYK-461reduces cardiac myocytes contractility by decreasing the adenosine triphosphatase activity and early, chronic administration of MYK-461 suppresses the development of HCM phenotype.
Teerlink JR, Diaz R, Felker GM, McMurray JJV, Metra M, Solomon SD, et al. Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N Engl J Med. 2020;384:105–16.
Toepfer CN, Garfinkel AC, Venturini G, Wakimoto H, Repetti G, Alamo L, et al. Myosin sequestration regulates sarcomere function, cardiomyocyte energetics, and metabolism, informing the pathogenesis of hypertrophic cardiomyopathy. Circulation. 2020;141:828–42.
Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106(6):1236–49.
Alamo L, Ware JS, Pinto A, Gillilan RE, Seidman JG, Seidman CE, et al. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife. 2017;6:e24634.
Cirino S, Lima FS, Gonçalves MB. Spatial distribution of specialized cardiac care units in the state of Santa Catarina. Rev Saude Publica. 2014;48(6):916–24.
Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358(18):1899–908.
Kaski JP, Syrris P, Esteban MTT, Jenkins S, Pantazis A, Deanfield JE, et al. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2(5):436–41.
García-Giustiniani D, Arad M, Ortíz-Genga M, Barriales-Villa R, Fernández X, Rodríguez-García I, et al. Phenotype and prognostic correlations of the converter region mutations affecting the β myosin heavy chain. Heart. 2015;101(13):1047–53.
Heitner SB, Jacoby D, Lester SJ, Owens A, Wang A, Zhang D, et al. Mavacamten treatment for obstructive hypertrophic cardiomyopathy a clinical trial. Ann Intern Med. 2019;170(11):741–8.
•• Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396(10253):759–69 Findings from this study suggest that treatment with mavacamten improves exercise capacity, LVOT obstruction, NYHA functional class, and health status in patients with obstructive HCM.
Kubo T, Kitaoka H, Okawa M, Yamanaka S, Hirota T, Baba Y, et al. Combined measurements of cardiac troponin I and brain natriuretic peptide are useful for predicting adverse outcomes in hypertrophic cardiomyopathy. Circ J. 2011;75(4):919–26.
Geske JB, McKie PM, Ommen SR, Sorajja P. B-type natriuretic peptide and survival in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;61(24):2456–60.
Seydelmann N, Liu D, Krämer J, Drechsler C, Hu K, Nordbeck P, et al. High-sensitivity troponin: a clinical blood biomarker for staging cardiomyopathy in fabry disease. J Am Heart Assoc. 2016;5(6):e002839.
Ho CY, Mealiffe ME, Bach RG, Bhattacharya M, Choudhury L, Edelberg JM, et al. Evaluation of mavacamten in symptomatic patients with nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2020;75(21):2649–60.
Saberi S, Cardim N, Yamani MH, Schulz-Menger J, Li W, Florea V, et al. Mavacamten favorably impacts cardiac structure in obstructive hypertrophic cardiomyopathy: EXPLORER-HCM CMR substudy analysis. Circulation. 2020;143(6):606–8.
Kazi DS, Bellows BK, Baron SJ, Shen C, Cohen DJ, Spertus JA, et al. Cost-effectiveness of tafamidis therapy for transthyretin amyloid cardiomyopathy. Circulation. 2020;141(15):1214–24.
Gurwitz JH, Maurer MS. Tafamidis-a pricey therapy for a not-so-rare condition. JAMA Cardiol. 2020;5(3):247–8.
Elliott PM. The end of the beginning for drug therapy in obstructive hypertrophic cardiomyopathy with EXPLORER-HCM. Cardiovasc Res. 2020;116(13):e175–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Olivotto is on the advisory board of EXPLORER-HCM trial and has received research funding and speakers’ fees from Myokardia.
The other authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Myocardial Disease
Rights and permissions
About this article

Cite this article
Zampieri, M., Argirò, A., Marchi, A. et al. Mavacamten, a Novel Therapeutic Strategy for Obstructive Hypertrophic Cardiomyopathy. Curr Cardiol Rep 23, 79 (2021). https://doi.org/10.1007/s11886-021-01508-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s11886-021-01508-0