
Abstract
Key message
SNP alleles on chromosomes 4BL and 6AL are associated with sensitivity to salt tolerance in wheat and upon validation can be exploited in the development of salt-tolerant wheat varieties.
Abstract
The dissection of the genetic and molecular components of salt stress response offers strong opportunities toward understanding and improving salt tolerance in crops. In this study, GWAS was employed to identify a total of 106 SNP loci (R2 = 0.12–63.44%) linked to salt stress response in wheat using leaf chlorophyll fluorescence, grain quality and shoot ionic (Na+ and K+ ions) attributes. Among them, 14 SNP loci individually conferred pleiotropic effects on multiple independent salinity tolerance traits including loci at 99.04 cM (R2 ≥ 14.7%) and 68.45 cM (R2 ≥ 4.10%) on chromosomes 6AL and 4BL, respectively, that influenced shoot Na+-uptake, shoot K+/Na+ ratio, and specific energy fluxes for absorption (ABS/RC) and dissipation (DIo/RC). Analysis of the open reading frame (ORF) containing the SNP markers revealed that they are orthologous to genes involved in photosynthesis and plant stress (salt) response. Further transcript abundance and qRT-PCR analyses indicated that the genes are mostly up-regulated in salt-tolerant and down-regulated in salt-sensitive wheat genotypes including NRAMP-2 and OPAQUE1 genes on 4BL and 6AL, respectively. Both genes showed highest differential expression between contrasting genotypes when expressions of all the genes within their genetic intervals were analyzed. Possible cis-acting regulatory elements and coding sequence variation that may be involved in salt stress response were also identified in both genes. This study identified genetic and molecular components of salt stress response that are associated with Na+-uptake, shoot Na+/K+ ratio, ABS/RC, DIo/RC, and grain quality traits and upon functional validation would facilitate the development of gene-specific markers that could be deployed to improve salinity tolerance in wheat.
Similar content being viewed by others

Introduction
Wheat is one of the world’s most important strategic food crops, with annual production of about 736 million metric tons (MMT) (FAO 2015). Due to the continuous decline in global production of major staple food crops since 1980 caused by global warming (Lobell et al. 2011), there is a need to increase wheat production amidst the projected increase in the world’s population (Jaggard et al. 2010) and the threat posed by continuous salinization of arable lands and ongoing climatic change. Over 6% of the world’s total land area is affected by salinity (FAO 2010). This is expected to increase in the coming years (Munns and Tester 2008), resulting in predicted US$12 billion annual losses in agricultural production (Qadir et al. 2008; Flowers et al. 2010). Soil salinization is an important land degradation problem in both dry and irrigated areas. Salinization can be reduced by leaching soluble salts out of the soil with excess irrigation water (Gupta and Goyal 2017), and/or high levels of soil salinity can be tolerated if salt-tolerant crops are grown (Ashraf et al. 2012). High salt concentration in soils reduces plant growth (Munns and Tester 2008; Zhang et al. 2010a) by limiting leaf growth and expansion (Cramer 2002). Salt-tolerant and salt-sensitive crops respond differently to salinity stress. The former tend to exclude excess salts (such as Na+) from the roots as well as compartmentalize the excess Na+ in leaf vacuoles to avoid damaging the photosynthetic machinery.
Salt stress leads to ionic imbalance and hyperosmotic stress in plants (Flowers 2004), causing severe damage to photosynthetic apparatus (Parida and Das 2005; Yamane et al. 2008) and reduction in seedling growth and survival rate (Lutts et al. 1995) which results in poor seed set and low crop yield (Asch et al. 2000; Atkinson and Urwin 2012; Nayidu et al. 2013). Grain quality is also adversely affected by salinity (Rao et al. 2013) due to the indirect effects of Na+ toxicity on the photosynthetic and biosynthetic processes within the chloroplasts. Under high salinity, the soil available K+ and Na+ compete for uptake in plant roots, and the degree to which one ion is preferred over the other determines adaptation and tolerance to salt stress. Zhang et al. (2010a) indicated that low K+- and high Na+-uptake are often correlated with each other. Thus, the identification of salt-tolerant genotypes possessing attributes such as reduced uptake of Na+, increased preference of K+ over Na+ in the root during uptake and translocation to shoots, increased tissue tolerance and production of enzymes, hormones, antioxidants, etc., is of great interest to wheat breeders. To meet future grain demands, breeders are required to develop new resource-efficient and salt-tolerant wheat cultivars by combining large-scale plant phenotyping with genome sequencing in forward genetics. Over the years, the traditional phenotyping methods have been used to evaluate crops agronomic and yield performance under salinity stress. However, these methods are labor-intensive, time-consuming and are considered to be one of the major bottlenecks in exploiting genetic information for genomic analysis (Rahaman et al. 2015) and advancement of yield improvement (Furbank and Tester 2011; McCouch et al. 2013). The evaluation of large populations in breeding programs has necessitated the search for traits linked to salinity tolerance that are economical, quick, and easy to measure; to facilitate the identification and mapping of salt tolerance (ST) QTL. The use of chlorophyll fluorescence (HTPchlF) techniques allows for non-invasive plant analyses and quick detection of stress or damage in the photosynthetic apparatus (Glynn et al. 2003; Chaerle et al. 2007). In turn, the potential in understanding common genetic factors regulating leaf photosynthetic activities, shoot K+ and shoot Na+ accumulation and grain quality in wheat would be very useful to breeders in bridging the gene to productivity/grain quality gap under salt-affected agro-ecologies.
Progress is being made in the discovery of quantitative trait loci (QTL) linked to ST genes involved in cell protection, transporter and oxidoreductase activities and regulatory elements which enhance the plant response to salt stress (James et al. 2006; Byrt et al. 2007; Huang et al. 2009; Zhang et al. 2012; Li et al. 2013, 2014a; Shi et al. 2014). The variable expressions of these genes are essential components of plant salt stress response and adaptation mechanisms and may be genotype specific and/or transcriptionally regulated by cis-regulatory elements (CREs). Several studies have revealed the association of DNA polymorphisms with ST in wheat (Genc et al. 2010; Xu et al. 2013; Masoudi et al. 2015; Turki et al. 2015), but are yet to be elucidated due to lack of functional and molecular validation. The understanding of how potential molecular cues relate to salt stress response remains a major challenge in molecular biology, as transcriptional mechanisms contribute to the regulation of nearly all cellular processes (Li et al. 2006; von Korff et al. 2009). Such understanding would facilitate exploiting them in genomic research and breeding programs to improve wheat salt stress tolerance and productivity. However, strengthening the connection between genotype and phenotype is a major challenge facing by plant molecular breeders (Pauli et al. 2016). The advances in next-generation DNA sequencing (NGS) have made identification of QTL/genes associated with complex agronomic traits possible. Genome-wide association studies (GWAS) provide dense genome coverage and allow for accurate identification of putative QTL in unstructured populations (Lorenz et al. 2011). It has been routinely used to identify the genetic basis of simple and complexly inherited agronomic and adaptive traits (Jighly et al. 2015; McCouch et al. 2016; Sun et al. 2017).
In this study, we used noninvasive high-throughput FluorPen FP100 to assess the leaf photosynthetic activities of a wheat diversity panel under saline and non-saline stress conditions. In addition, shoot K+ and Na+ accumulation and grain quality (GQ) under salt stress conditions were also assessed. The objectives of this study were to: (i) identify HTPchlF traits that may serve as possible surrogates for screening salt stress response in wheat, (ii) identify QTL linked to ST using the HTPchlF, shoot ionic and GQ traits and (iii) annotate functionally the associated SNP loci to uncover the putative genes involved and gain insights into their salt response mechanisms by in silico protein, expressions, promoter and expressed sequence analyses. To our knowledge, this is the first study that simultaneously evaluated and analyzed the genetic architectures of salt stress response in a wheat diversity panel using three independent parameters: leaf chlorophyll fluorescence, shoot Na+/K+ accumulation and grain quality.
Materials and methods
Plant material and growth conditions
The 150 wheat genotype diversity panels including the hydroponic screening systems used in this study have been described previously in Oyiga et al. (2016). Briefly, the germplasm was grown in three replicated hydroponic systems in the greenhouse between September and December 2016 under non-saline (control) and 150 mM NaCl (13.24–14.71 dS m−1) saline conditions. The experiment was repeated two times. Salt stress was initiated 3 days after planting by addition of 50 mM NaCl incrementally for 3 days. After this time, the full 150 mM NaCl treatment was sustained for 24 days until harvest.
Leaf chlorophyll a fluorescence
At day 24 of salt stress treatment, three readings on the HTPchlF traits were taken at three positions on the blade of the third leaf (c. 5 cm from the stem, toward the centre, and c. 5 cm from the tip) using the FluorPen FP100 (Photon Systems Instruments, Brno, Czech Republic). Readings were taken on the light-adapted leaves of six plants per genotype. In total, 18 data points per genotype were used for analyses. Light intensity reaching the leaves during the measurement was set at 3000 mol (photons) m−2 s−1, which was sufficient to generate maximal fluorescence. The HTPchlF parameters evaluated (Strasser et al. 2004) are described in Table 1.
Shoot K+ and Na+ accumulation during salt stress
The amount of Na+ and K+ (in g/100 g sample DW) accumulated in the shoot (leaf parts plus stem) after 24 days of salt treatment was measured using atomic absorption spectrophotometer (type 2380; PerkinElmer, Wellesley, MA, USA), as described in Oyiga et al. (2016), followed by the calculation of their corresponding K+ to Na+ ratios.
Determination of the grain quality (GQ)
The GQ traits including grain protein content (GPC), grain starch content (GSC), grain neutral detergent fiber (NDF) and grain crude fiber (CFC) of the 150 wheat diversity panel were also measured from grains harvested from three replicated field trials in Karshi, Uzbekistan, under high-saline (9.24–17.58 ds/m) and low-saline (2.40–6.34 ds/m) field conditions. The GQ analysis was performed using a DA 7250 NIR analyzer (PerkinElmer, Wellesley, MA, USA).
Statistical analyses of the phenotype data
Due to the positioning (row and column) effects in the experiments, all the traits evaluated were analyzed following the restricted maximum likelihood (REML) procedure as implemented in GENSTAT 16. The plant position effects were accounted for by including “Replication/Row*Column”—rows crossed with columns nested within replication as sources of random effects in REML (Payne et al. 2015). Statistically significant differences were ascertained with the Wald’s test. Variance components due to genotypic (\(\sigma_g^2\)) and environmental (\(\sigma_e^2\)) effects were determined [by considering genotypes as random effect and, by taking into account the genotype by salt-treatment interaction (Piepho et al. 2008)] and were used to estimate the traits broad-sense heritability (h2) as implemented in GENSTAT 16 for REML (O’Neill 2010) as: \(h^2 = \sigma_g^2/(\sigma_g^2 + \sigma_{gT}^2/T + \sigma_R^2/r)\), where \(\sigma_{gT}^2\) is genotype × salt-treatment interactions variance, T is the number of number of treatments, \(\sigma_R^2\) is the residual variance and r is the number of replications in each treatment. Best linear unbiased predictors (BLUPs) were used to calculate the salt tolerance indices (STIs) of all the traits, as described in Oyiga et al. (2016). The STI estimates were included in the GWAS model as phenotypes for the localization of QTL associated with salt stress response in the diversity wheat panel. Correlation coefficients (r) for each pair of STI trait were also obtained using the SPSS 16.0 program for windows.
Genetic analysis of the 150 wheat diversity panel
Detailed information of SNP genotyping, population structure (PS) and linkage disequilibrium (LD) analyses of the diversity panel have been described in Oyiga et al. (2018). A subset of 18,085 polymorphic SNP markers remaining after data cleaning (missing values > 5% and minor allele frequency, MAF < 5%) were used for GWAS analysis which was performed with two software programs: TASSEL 5.2.13 standalone version (Bradbury et al. 2007) and SAS (SAS Institute Inc., Cary, NC). Only the congruent SNP loci identified by both programs were reported after employing the mixed linear model (MLM) adjusted for PS (Q matrix, as the fixed covariate) and kinship (K-matrix, as the random effect) matrixes. The cutoffs for accepting significant marker–trait associations (MTA) were calculated according to Long et al. (2013): -log10 (α/#tests), where α = 0.05 and #tests = the number of effective tests calculated as the total genome coverage divided by genome linkage disequilibrium (LD). Based on LD of the population, all SNP loci detected at genetic intervals defined by the genome LD of 10 cM for A-genome, 11 cM for B-genome and 14 cM for D-genome (Oyiga et al. 2018) were considered to be in high LD (Breseghello and Sorrells 2006; Pasam and Sharma 2014) and were grouped in one SNP cluster. Thereafter, the principal-coordinate analyses (PCoA) based on all the detected SNP loci were performed in GenAlEx 6.5 (Peakall and Smouse 2012) using the salt-tolerant and salt-sensitive wheat genotypes that were identified in the diversity panel (Oyiga et al. 2016), to check their genetic diversity for salt stress tolerance.
In silico identification and ontological analysis of the detected SNP loci
Based on SNP marker sequence information, candidate genes containing associated SNP markers were identified using NCBI BLAST (www.ncbi.nlm.nih.gov/BLAST), as previously described in Oyiga et al. (2018).
Gene expression analyses of candidate genes harboring the associated SNP loci
The third leaves harvested from 4-week-old Altay2000 and Bobur after 2 h, 11 days and 24 days under saline (100 mM NaCl) and non-saline conditions were used for expression analyses of the candidate genes. Altay2000 and Bobur are among the four wheat genotypes—Altay2000 and UZ-11CWA-8 (salt-tolerant) and UZ-11CWA-24 and Bobur (salt-sensitive), which showed consistent and contrasting response to salinity stress across three different developmental growth stages identified in the GWAS panel assessment (Oyiga et al. 2016). The expression level of the candidate genes after 24 days of salt stress was included because it coincided with the time period at which the GWAS phenotypic data for both HTPchlF and shoot K+- and Na+-accumulation were collected. The blades of the leaf samples from five plants per genotype were pooled and analyzed using the quantitative next-generation sequencing (NGS) based on the massive analysis of cDNA ends (MACE) transcriptome profiling.
RNA isolation and RT-qPCR of the associated candidate genes
Leaf samples from the third leaf of three plants of Altay2000 and UZ-11CWA-8 (salt-tolerant), and UZ-11CWA-24 and Bobur (salt-sensitive) wheat genotypes were collected after 30 days in both saline (100 mM NaCl) and non-saline conditions, flash frozen in liquid nitrogen and preserved at − 80 °C until use. Total RNA was extracted from the harvested leaves using the E.Z.N.A. Plant RNA Kit (Omega Bio-Tek, Norcross, GA, USA), followed by DNA digestion with the Omega Bio-Tek DNA Digestion kit. The qualities of the extracted RNA samples were verified with electrophoresis (1.5% agarose gel) and quantified spectrophotometrically using NanoDrop 2000c (PEQLAB Biotechnologie GmbH, 6404 Polling).
The cDNA synthesis was performed with first-strand cDNA Synthesis kit—Cat.#K1632 (ThermoFisher Scientific, Massachusetts, USA). Primer pairs (Table 2) were designed for OPAQUE1 (myosin-J heavy chain) on 6AL, ABC transporter F family member 3 (TaABCF3) on 6AL and NAD(P)H-quinone oxidoreductase subunit L (NAD(P)H) on 5AL using the online Primer3 program (http://primer3.wi.mit.edu/). Gene amplification via RT-qPCR was carried out in biological triplicates of 20 μl final volume reactions containing 3 μl of cDNA template, 0.3 μl of each gene-specific primer, 10 μl of DyNAmo ColorFlash SYBR Green 2X-master mix with ROX- Cat.#F456L (ThermoFisher Scientific, Massachusetts, USA) and 6.4 μl RNase-free water, with the following cycling conditions: pre-denaturation 95 °C/7 min and then for 40 cycle: 95 °C/10 s, 60 °C/30 s, 72 °C/30 s. Gene amplification and fluorescence data acquisition were conducted on SDS-7500 Sequence Detection System (Applied Biosystems, USA) using cDNA obtained from the stressed and non-stressed salt-tolerant and salt-sensitive plants. The expressed gene transcript amounts were normalized with TaEf-1a and TaEf-1b (Unigene accession: Ta659; Table 2), following the Livak and Schmittgen (2001) method.
In many cases, the associations identified in GWAS may provide the initial landmarks for the identification of the candidate genes underpinning genetic variants and do not represent the causal effect but rather one in LD; the scaffolds corresponding to the genetic intervals (in LD) of the pleiotropic SNP loci detected at 68.45 cM and 90.04 cM on 4BL and 6AL chromosomal regions, respectively, were identified and analyzed using the JBrowse (Skinner et al. 2009) in the International Wheat Genome Sequencing Consortium database (IWGSC, https://www.wheatgenome.org). Scripts written in R program were used to identify all the functionally annotated candidate genes (iwgsc_refseqv1.0_FunctionalAnnotation_v1__HCgenes_v1.0-repr.TEcleaned.TAB) in both intervals, and their transcript abundances obtained from the MACE data of salt-tolerant (Altay2000) and salt-sensitive (Bobur) wheat genotypes under saline and non-saline conditions were analyzed.
Promoter region analyses of three candidate gene targets identified
Because CREs regulate the transcription of the neighboring genes, promoter regions of three identified salt stress-responsive candidate genes including NRAMP-2, TaABCF3 and CSLF1 were analyzed in the two salt-tolerant (Altay2000 and UZ-11CWA-8) and two salt-sensitive (UZ-11CWA-24 and Bobur) wheat genotypes for identification of possible regulatory elements for salt stress tolerance in wheat. Specific oligonucleotides for: NRAMP-2 (5′-TCCGGGGCGTTGCTTGAA-3′; 5′-TCGGAGATGGAGATGGAGACCT-3′; Ta = 60 °C, TaABCF3 (5′-CAACCAACTAGAAGCGAGTG-3′ and 5′- GGCGATGTAGGAGACGAT-3′; Ta = 59.0 °C) and CSLF1 (5′- TGTACATAGCGTCCAGATTTG-3′ and 5′-ATCGTCATTTCACCAGCAAC-3′; Ta = 55.5 °C) were designed and used for gene fragment amplification and sequencing. The nucleotide sequences of the amplified candidate gene targets were compared and aligned with each other using MotifViz (Fu et al. 2004; http://biowulf.bu.edu/MotifViz) and PlantPAN 2.0 (Chow et al. 2015; http://PlantPAN2.itps.ncku.edu.tw/). In addition, the amino acid sequences of the analyzed candidate genes at coding regions were compared with their corresponding draft sequences obtained from the Ensembl Web site (Kersey et al. 2016), to check for possible allelic variations that may cause structural and functional variations during salinity stress, using MAFFT version 7 (http://mafft.cbrc.jp/alignment/server/) and Sequence Manipulation Suite (Stothard 2000).
Results
Phenotypic analysis of the wheat diversity panel
The effects of salt stress on photosynthetic-related and grain quality traits were analyzed via high-throughput phenotypic methods. Substantial genetic variation was observed in all the HTPchlF traits investigated, as reflected by their corresponding mean, standard deviation (SD) and genotypic coefficient of variation (GCV) values under saline and non-saline conditions (Table 3). The GCVs (GCV = √Genetic variance/Average × 100%) ranged from 0.9 in Fv/Fm to 13.67% in PI(ABS) under non-saline conditions and from 1.37 in Fv/Fm to 16.40% in PI(ABS) under saline conditions. The skewness and kurtosis values indicated that HTPchlF traits were more or less continuously distributed and thus considered to be quantitatively inherited. REML results showed that variation among genotypes and salt stress treatments were significant (P < 0.01) on the HTPchlF traits. Application of salt stress adversely affected the HTPchlF traits by decreasing their values from − 1.43% in Fv to − 37.04% in PI(ABS), except for DIo/RC (+ 9.71%) and ETo/RC (+ 8.98%). Further, HTPchlF traits were heritable with h2 estimates which ranged from 19% in Fv to 63% in ETo/RC, indicating that the observed genotypic differences can be used for mapping ST QTL.
To characterize the effect of salinity on GQ, several grain parameters were defined. All the evaluated GQ traits (except NDF) were strongly influenced by genotype, salt stress and their interaction (Table 3). Substantial variation that could allow for genetic characterization of salt stress responses was observed in GQ traits. The GCVs were: 9.00% and 7.21% for GPC, 8.03% and 8.44% for CFC, 5.33% and 6.19% for NDF, and 1.52% and 1.56% for GSC under non-saline and saline conditions, respectively. The GQ traits followed a continuous distribution in both saline and non-saline-stress field conditions. Field salinity increased GPC by 13.05% and NDF by 1%, but decreased the GSC and CFC by 1.2% and 1.3%, respectively, while h2 estimates for QG traits were highest in CFC (98%), followed by GSC (93%), GPC (89%) and NDF (87%).
The majority of the STI estimates of HTPchlF traits were significantly correlated (P ≤ 0.01) among each other (Table 4), with the strongest correlations (r) found between Fv/Fo and Fv/Fm (r = 0.87**), ABS/RC and DIo/RC (r = 0.83**), and Fv/Fo and PI(ABS) (r = 0.82**). The correlation values among GQ traits indicated a large but negative correlation between GPC and GSC (r = − 0.80**). In addition, the shoot K+/Na+ ratio showed moderate (P ≤ 0.01) correlation with ABS/RC, TRo/RC, Fv, Fv/Fo and PI(ABS). Similarly, Fv correlated negatively and positively with GPC (r = − 0.24**; P ≤ 0.01) and GSC (r = 0.26**; P ≤ 0.01), respectively.
Identification of QTL for ST in the diversity panel
The goal was to identify chromosomal regions and loci harboring genetic factors associated with salt stress response among the 150 wheat genotype diversity panel. Results identified a total of 106 SNP loci linked with STIs of HTPchlF, shoot ionic and GQ ST traits (Fig. 1; Table S1), and they explained between 0.12 and 63.44% of the observed phenotypic variance (PVE). The PCoA performed based on the detected genetic variants accurately separated salt-tolerant and salt-sensitive wheat genotypes into two groups (Fig. 2). Salt-tolerant genotypes were mostly positioned on the left, whereas salt-sensitive lines were distributed on the right side of the plot. The first three PCos accounted for 28.97% of the PVE, and the major genetic contributors to the observed PVE for salt stress response were detected on chromosomes 1AL (at 141.53–144.94 cM), 2BL (at 155.41 cM), 2DL (at 97.42–105.13 cM), 4AS (at 43.39 cM), 4BL (at 68.45 cM), 6AL (at 99.04 cM) and 7AL (at 126.8 and 148.43 cM).

Genetic map of wheat showing the location of the genetic variants (SNP markers) associated with the chlorophyll fluorescence (in green), shoot K+ and shoot Na+ (in blue) and seed quality (in red) salt tolerance traits in the 150 wheat genotype diversity panel. Each bar denotes the associated chromosomes, and the black-shaded regions are the associated chromosomal regions (SNP clusters) as defined by the genome LD. The QTL names for each SNP cluster have been presented in the left side the chromosomes

Principal coordinates analysis (PCoA) plot using a genetic distance matrix (GenAlEx 6.5) estimated with data from 115 associated polymorphisms of the salt-tolerant (black color/triangular shaped) and salt-sensitive (red color/squared shape) wheat genotypes previously identified among the studied population. The genotypes (in bold) were used to perform the gene expression and promoter region/cis-element analyses
To investigate the pattern and the likely causal relationship among the traits with respect to salt stress response, the effect of all linked SNP loci within each LD-defined genetic interval “SNP clusters” was considered to be emanating from single or few genes in high LD. Thus, the 106 associated loci were resolved into 16 unique SNP clusters across the wheat genome, out of which 10 showed strong genetic association with at least two of the independent ST traits evaluated (Table 5; Fig. 1). The SNP cluster on 2BL (PVE = 3.5–8.3%) which spanned an interval of 1.34 cM strongly influenced ST in Fv/Fm, shoot Na+ uptake, shoot K+/Na+ ratio and GPC ST traits. Four SNP clusters, notably on chromosomes 1AS, 3AS, 5AS, and 6BL, were linked with HTPchlF and GQ ST traits, while an additional four SNP clusters on 5BL and 6AL strongly influenced both the HTPchlF and shoot ionic (S +K – +Na ) ST traits. Previous studies have shown that several SNP clusters detected in this study are in genetic regions known to carry salt stress response QTL (Table 5), while novel QTL regions (“SNP clusters”) were also identified.
Single SNP loci showing genetic effects on multiple ST traits
The goal was to identify SNP loci exhibiting pleiotropic effects on multiple ST traits. We anticipated that the network of traits sharing genetic association to common SNPs and/or genes may offer some clues as to the underlying shared genetic and molecular mechanisms of salt stress response in wheat. Results from this study showed that a total of 14 SNP loci conferred pleiotropic effects on several ST phenotypes (Table 6). Among them is the locus at 90.04 cM (PVE ≤ 14.7%) on chromosome 6AL which exhibited the most influence with the highest number of ST traits: ABS/RC, DIo/RC, shoot Na+ uptake and shoot K+/Na+ ratio. Similarly, the SNP locus at 99.80 cM (PVE ≤ 8.3%) on 2BL controlled salt stress response of three ST traits: Fv/Fm, CFC, and shoot Na+ uptake, while the SNP locus at 82.38 cM (PVE ≤ 14.9%) on chromosome 6AL had a strong effect on the ST traits for specific energy fluxes (per reaction center) for energy absorption (ABS/RC) and dissipation (DIo/RC), and shoot Na+ uptake. In addition, the locus at 118 cM on 7AL affected DIo/RC, Fv/Fm, and shoot K+/Na+ ratio, whereas the SNP locus at 71.76 cM (PVE ≤ 4.1%) on chromosome 6BL influenced only GQ traits: NDF, CFC, and GPC. A locus at 68.45 cM on chromosome arm 4BL associated with three ST traits including specific energy fluxes (per reaction center) for energy absorption (ABS/RC) and trapping (TRo/RC) and shoot K+/Na+ ratio. Further analysis indicated that most of the SNP markers with pleiotropic effects on the ST traits were located in the coding regions of known photosynthetic and abiotic/salt stress-related candidate genes (Table 6). The presence of SNPs and sequence variations in the gene coding region can influence gene structure and function resulting in altered plant responses to stress.
Annotation and ontological classifications of the associated SNP loci
Most of the putative candidate genes identified belong to categories of genes involved in abiotic stress and salt stress response. They include those involved in stress response (27.9%), ion/metal transport (12.8%), carbohydrate and energy metabolisms (10.5%) and photosynthetic (10.5%) activities. Others include genes in high sequence homology with the SNP markers identified that function in the protein repair maintenance (10.5%) and transcription factor (10.5%) pathways. Few of the SNP marker sequences identified coded for genes involved in reactive oxygen species (ROS) scavenging (4%) and protein translation (2%) activities. The ontological analyses of the mapped genetic variants provided evidence that the SNP loci identified are coding for salt stress response (data not shown).
The SNP marker at 90.04 cM that exhibited pleiotropic effects on ABS/RC, DIo/RC, shoot Na+ uptake and shoot K+/Na+ ratio on 6AL is homologous to OPAQUE1 that encodes a Myosin XI motor protein (myo), while the locus at 99.80 cM on 2BL showed high sequence homology to putative mixed-linked glucan synthase 3. The SNP marker at 82.38 cM on 6AL associated with ABS/RC, DIo/RC and shoots Na+ uptake showed a high sequence identity with universal stress protein A-like protein (USP). The SNP locus at 71.76 cM on 6BL closely linked to NDF, CFC and GPC and was located within the domain of Galactoside 2-alpha-L-fucosyltransferase, while the LRR receptor-like serine/threonine-protein kinase transcript was identified at 118 cM (which is strongly influenced DIo/RC, Fv/Fm, shoot K+/Na+ ratio) on 7AL. Additionally, the SNP marker on 6AL at 140.87 cM associated with Fv and shoot Na+ uptake showed high sequence identity with TaABCF3 transporter. The SNP markers at 68.45 cM on 4BL associated with shoot K+/Na+ ratio, ABS/RC, and TRo/RC revealed high sequence identity with NADH dehydrogenase complex (plastoquinone) assembly (NRAMP-2) involved in the photosynthetic electron transport in photosystem I and metal ion transporter/homeostasis. The biological functions of these candidate genes are presented in Table 6.
Expression analyses of genes associated with the detected ST SNP marker
To gain understanding whether the underlying candidate genes are responsive to salt stress, we investigated the expression levels and the dynamics of 28 associated gene transcripts in the third leaves of Altay2000 (salt-tolerant) and Bobur (salt-sensitive) wheat genotypes for up to 24 days under saline (100 mM NaCl) and non-saline conditions. The transcript amounts were obtained from genome-wide gene expression profiling performed using the MACE transcriptome approach. As shown in Table 7 and Fig. 3, the analyzed candidate genes showed differential expression between the salt-tolerant and salt-sensitive wheat genotypes. Except in few instances, all the genes were up-regulated and/or highly expressed (relative to the non-saline condition) in Altay2000 and down-regulated in Bobur. For instance, OPAQUE1 was up-regulated by +198.0% in Altay2000 and + 32.9% in Bobur, while NRAMP-2 was also up-regulated by + 321.5% in Altay2000 and down-regulated by − 65.8% in Bobur. The activity of the TaABCF3 transporter increased by + 47.7% and + 2.9% in the salt-tolerant and salt-sensitive wheat genotypes, respectively, and sucrose-phosphate synthase increased by + 231.9% in Altay2000 and + 35.2% in Bobur.

Effect of salt stress on some of the identified gene transcript abundance (% change to control) between salt-tolerant genotype (Atlay2000, in black) versus salt-sensitive genotype (Bobur, in gray) after 24 days of stress. OPAQUE1 = Myosin-J heavy chain; Os03g0405500 = Probable nucleoredoxin 1-1; IAA13 = Auxin-responsive protein; hxB = Molybdenum cofactor sulfurase; cnb-1 = Calcineurin subunit B; SPS1 = Probable sucrose-phosphate synthase 1; pkiA = Pyruvate kinase; USP = UDP-sugar pyrophosphorylase; CSLF1 = Putative mixed-linked glucan synthase 1; JMJ703 = Lysine-specific demethylase; MPK9 = Mitogen-activated protein kinase 9; TRIUR3_02773 = Putative serine/threonine-protein kinase Cx32, chloroplastic; CRS1 = Chloroplastic group IIA intron splicing facilitator; TaABCF3-un = ABC transporter F family member 3 protein; kefC = Glutathione-regulated potassium-efflux system protein; TRIUR3_16707 = Multiple C2 and transmembrane domain-containing protein 1; TaABCE2 = ABC transporter E family member 2, NRAMP2 = NADH dehydrogenase complex (plastoquinone) assembly (Metal transporter- Nramp2); HAK10 = Potassium transporter 10
The salt stress response kinetics of Altay2000 and Bobur at 2 h, 11 days and 24 days under saline stress and non-saline conditions were analyzed using sigmoidal nonlinear statistics (Figure S1). Results indicated that the temporal patterns of the expressed transcripts change over time during salt stress exposure and varied between the salt-tolerant and salt-sensitive wheat genotypes. Altay2000 and Bobur exhibited differential expression signatures at 2-h stress time period, with the latter showing mostly higher transcript amounts. The observed trends were maintained for few (0–5 days) days and thereafter changed at 11 days of salt stress with the transcript amount induced by salinity in Altay2000 rising steadily and exponentially and mostly decreased in Bobur. Further, genes such as OPAGUE1, molybdenum cofactor sulfurase (hxB) and NRAMP-2 exhibited an early (0–5 days) salt stress response, while mitogen-activated protein kinase 9 (MPK9) was differentially expressed after 15 days of salt stress application.
The RT-qPCR performed to analyze the gene expression patterns after an extended period of 30 days under salt stress also revealed increased activities of OPAQUE1, TaABCF3-un and NAD(P)H-quinone oxidoreductase subunit L, chloroplastic in the two salt-tolerant (Altay2000 and UZ-11CWA-8) genotypes, compared with the two salt-sensitive (UZ-11CWA-24 and Bobur) genotypes (Fig. 4). The candidate gene OPAQUE1 was highly expressed in the salt-tolerant genotypes, with no corresponding and meaningful transcriptional changes in the salt-sensitive genotypes. However, salt stress induced greater transcriptional changes in TaABCF3-un and NADPH in both the two salt-tolerant and two salt-sensitive wheat genotypes, although the fold-changes were much higher in the two salt-tolerant genotypes.

The qRT-PCR analyses of OPAQUE1 (on 6AL): myosin-J chain, TaABCF3-un (on 6AL): ABC transporter F family member 3 and NADPH (on 5AS): NAD(P)H-quinone oxidoreductase subunit L, chloroplastic in leaves of two salt-tolerant (Atlay2000 and UZ-11CWA-8) and salt-sensitive (UZ-11CWA-24 and Bobur) after 30 days in non-saline (gray) and saline (black) conditions. Efa1.1 and Efa1.2 genes were used as internal control genes. Bars are the means ± standard error (n = 3)
Identification and expression analyses of the genes in the interval surrounding the detected SNP markers
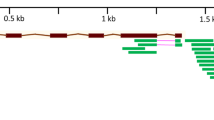
The Blast search in the IWGSC database showed that the genetic interval in LD block within the scaffold41600 (~ 13 Mb containing 61 ORFs or putative genes) on 4BL contains the SNP marker located at 68.45 cM (Table S2; Fig. 5), whereas the interval covering ~ 12 Mb in scaffold126294.1 contains 141 candidate genes and corresponds to the chromosomal region containing the pleiotropic SNP marker (at 90.04 cM) on 6AL (Table S3; Fig. 6). Results from this study indicated that TraesCS4B01G254300.1 which codes for NRAMP-2 was the most highly differentially expressed gene between the salt-tolerant and salt-sensitive genotypes after 24 days when the expressions of all the genes in the associated interval in scaffold41600 were analyzed. Salinity stress increased the expression of TraesCS4B01G254300.1 by + 363.4% in Altay2000 but decreased it by − 73.49% in Bobur. Highest transcriptional change differentials were also observed between the salt-tolerant (+ 3524.72%) and salt-sensitive (+ 734.90%) in TraesCS6A01G334400.1 (tyrosine kinase proteins) which is 317 bp away from TraesCS6A01G336500.1 (coding for OPAQUE1) that contains the SNP locus with pleiotropic effects on ST traits detected on chromosome 6AL.

Relative expression levels of all the annotated genes (in their natural order) belonging to scaffold41600 (~ 13 Mb) due to salt stress in Altay2000 (salt-tolerant) and Bobur (salt-sensitive) genotypes after 24 days. The relative abundance of the genes was determined as a change in transcript amounts after 24 days of salt stress relative to an untreated control. The “thick blue arrow” represents the gene domain carrying the GWAS mapped pleiotropic SNP marker sequences

Relative expression levels of all the annotated genes (in their natural order) belonging to scaffold126294.1 (~ 12 Mb) due to salt stress in Altay2000 (salt-tolerant) and Bobur (salt-sensitive) genotypes after 24 days. The relative abundance of the genes was determined as a change in transcript amounts after 24 days of salt stress relative to an untreated control. The “thick blue arrow” represents the gene domain carrying the GWAS mapped pleiotropic SNP marker sequences
Promoter response elements of the underlying salt-tolerant genes
To shed light into the mechanisms of the underlying gene expression regulations, the promoter regions of NRAMP-2, OPAQUE1, CSLF1 and TaABCF3 candidate genes were examined for possible detection of potential cis-elements involved in salt stress response using the salt-tolerant and salt-sensitive genotypes (Figures S2–4). The two tolerant genotypes showed unique DNA-binding domains of TF_motif_seq_0239/Dof (5′-AA[AG]G-3′) and PBF (5′-AAAGC-3′) at 99 bp upstream of the NRAMP-2 start codon (Figure S2). However, these motif features were found to be lacking in the two sensitive genotypes but instead were replaced by the broad-complex 3 (5′-TCAACAAAAAC-3′)—and/or hunchback (5′-CAACAAAAAC-3′)—DNA-binding domains. The promoter sequence of 950-bp upstream of the OPAQUE1 gene start codon revealed no differences in the transcription factor binding sites of the contrasting ST wheat genotypes (Data not shown). The analysis of CSLF1 promoter regions showed genotype-specific motif variation among the wheat genotypes including the homeodomain motif family—bZIP; HD-ZIP (5′-GCAATCATAG-3) and GATA-binding proteins (5′-GATA-3) identified at 581 and 554 bp, respectively, upstream of the gene start codon of UZ-11CWA-08; the SPI-B (5′-TTCCGCT-3′) and NF-Y (5′-ATGAACCAATGCATGC-3′) found at positions 618 bp and 540 bp of UZ-11CWA-24, respectively (Figure S3). Motif differences were not detected among the genotypes in TaABCF3; however, the translation initiator codon of the gene was 21-bp downstream in UZ-11CWA-24 when compared to the other genotypes (Figure S4).
Structure analysis of the gene coding regions
Part of the objective of this study was to identify allele sequence differences between salt-tolerant and salt-sensitive genotypes within the candidate genes encompassing the detected pleiotropic SNP loci since their residue sequence pattern and constitution may affect the protein turnover as well as alter the underlying gene structure and function. Comparative sequence analysis revealed several point polymorphisms; some of them were non-synonymous when Altay2000 and Bobur were compared (Fig. 7). Four SNP sites were detected in exon 37 of OPAQUE1 (Fig. 7a), leading to the following putative changes: Cysteine to Glycine at position 1529 (C1529G), Alanine to Valine at position 1549 (A1549V), Arginine to Glycine at position 1626 (R1626G) and Cysteine to Tryptophan at position 1628 (C1628W). Several polymorphic sites which may have altered the structure and functions of NRAMP-2 (Fig. 7b), Dihydroorotate dehydrogenase (quinone) (Fig. 7c) and UDP-glucose 6-dehydrogenase 2 (Fig. 7d) were also detected with the mapped SNP loci.

Comparison of the deduced EST amino acid sequence in Atlay2000 (salt-tolerant) and Bobur (salt-sensitive) genotypes with their corresponding draft sequence obtained from Ensembl Genomes database (http://www.ensemblgenomes.org). Black and white colors indicate the identical and mutation sites, respectively, while gray colors represent the regions anchoring the core sequences of the SNP marker identified in this study
Discussion
Developing salt-tolerant cultivars is the preferred breeding strategy for improving wheat adaptation to salinity stress. This strategy requires the comprehensive understanding and identification of genetic and molecular mechanisms involved in salt stress response in wheat. Previous studies indicated that the studied wheat diversity panel responded differently to salt stress when yield-related traits across three growth stages and the shoot Na+ content and shoot K+/Na+ ratio of the third leaves were evaluated (Oyiga et al. 2016, 2018). The main goal of this study was to explore genetic variation in the diversity panel to identify genomic regions and candidate genes involved in salt stress response by analyzing simultaneously different plant physiological and grain quality parameters under different salt stress regimes. To the best of our knowledge, this is the first study that simultaneously evaluated the genetic mechanisms of salt stress response in wheat using three independent salt stress parameters. The identification of genetic variants that have pleiotropic effects on the ST traits would support further molecular breeding research efforts toward improving wheat adaptation and tolerance to salinity stress.
Phenotypic variability among the 150 diversity panel
The studied panel showed substantial (P < 0.001) phenotypic variation for salt stress tolerance in all the HTPchlF, GQ and shoot ionic traits evaluated (Table 2) with moderate-to-high h2 estimates, suggesting that the traits evaluated can be exploited to uncover the genetic architecture governing salt stress response in wheat.
Application of salt stress impacted negatively on the HTPchlF traits including the maximum quantum yield of PSII chemistry (Fv/Fm) and specific energy fluxes (per reaction centre) for both absorption (ABS/RC) and trapping (TRo/RC), indicating that the photosynthetic activities of the evaluated germplasm were suppressed under salinity stress. It has been shown that salt stress reduces photosynthetic activities in plants via the inhibition of photosystem II complex (PSII) at both acceptor [QA] and donor sides (oxygen-evolving complex OEC), and destruction of chlorophyll pigments by the accumulation of toxic ions (Chen and Murata 2011; Shu et al. 2012). The reduction observed in Fv/Fm may be associated with damage to the PSII reaction center (Kadir et al. 2007) and the decrease in the quantum efficiency of PSII photochemistry (Mathur and Jajoo 2014). The dissipation energy (DIo/RC) and electron flux (ETo/RC) per active reaction center increased during salt stress, due to the increased proportion of the inactive reaction centers in the leaves exposed to salt stress (Mehta et al. 2011), resulting in the ineffective exploitation of light energy received in the inactivated reaction centers (Satoh et al. 1983). Demetriou et al. (2007) and Mathur et al. (2013) have shown that the increase in ETo/RC in plant leaves during stress is related to the increasing number of inactive centers and the inefficient transfer of electrons from QA to QB. The performance index of PSII-PI(ABS), which is the combination of the indexes of ABS/RC, phi_Po and psi_0 (Appenroth et al. 2001), decreased under salt stress supporting the earlier results that PSII activities are affected by salt stress.
The analyses of GQ traits have shown that GPC and GSC are increased and decreased, respectively, under field salinity stress. Reports showing the impact of major abiotic stresses on GQ traits abound, but none have reported on effect with salinity stress in wheat. For instance, ozone (O3) (Piikki et al. 2008; Zheng et al. 2013) and heat (Farooq et al. 2011) stresses have been shown to increase the GPC in wheat, while the study conducted by Fuhrer et al. (1990) revealed that O3 stress decreases the GSC. In rice, the GSC decreased under salinity stress but the GPC increased (Baxter et al. 2011; Thitisaksakul et al. 2015), which is consistent with the findings from this study, implying that salinity stress increases the ability of plants to mobilize N in developing seeds at the expense of the carbon sink and source, resulting in less dry matter accumulation. The decrease in GSC might be connected with the lower plant growth rate—a direct consequence of the photosynthetic limitation in plants, which may have indirectly contributed to a reduction in the sink capacity and less starch deposition in the grains.
Correlations among ST traits
Correlated traits are of interest in plant breeding because they help to: (i) connect the genetic causes of the pleiotropic action of genes, (ii) understand how selection for one character will produce a simultaneous change in another character, and (iii) determine the relationship between traits and fitness (Sandhu et al. 2013). Our results indicated that the Pearson’s correlation coefficients (r) among the salt tolerance indices of HTPchlF trait were in many cases statistically significant, indicating that the salt stress responses via the pathways leading to photosynthesis follow similar pattern. Positive correlations were observed between shoot K+/Na+ ratio and some HTPchlF trait indices including Fv, ABS/RC, TRo/RC and Fv/Fo. High uptake of K+ improves plant stomatal movement, energy transfer and photosynthetic activities (Marschner and Marschner’s 2012), while the high shoot K+/Na+ ratio is essential for ion homeostasis in salt-stressed plants (Maathuis and Amtmann 1999; Cuin and Shabala 2006; Volkov and Amtmann 2006). Using these correlated HTPchlF traits as a “physiological marker” for indirect measurement of shoot K+/Na+ ratio (ion homeostasis) may offer an indirect and rapid approach for evaluating and mapping QTL linked to salt stress responses, as proposed by Li et al. (2014b). The negative correlation (r = − 0.82; P < 0.01) observed between GPC and GSC in this study has also been reported in wheat by Hucl and Chibbar (1996) and Burešová et al. (2010). Our findings suggest that the correlated HTPchlF traits have a great potential for application in wheat-breeding programs focused on accelerating the QTL mapping and incorporation of ST alleles into cultivated varieties. Together these shorten the generation interval and increase accuracy of selection to reduce the cost of breeding.
Genome-wide association studies (GWAS)
The genome-wide scan identified 106 SNP markers (PVE = 0.12–63.44%), belonging to 16 LD-defined SNP clusters, associated with ST in the diversity wheat panel. Among them, 60, 23 and 23 SNP markers were associated with HTPchlF, shoot ionic (shoot K+ uptake, shoot Na+ uptake and shoot K+/Na+ ratio) and GQ parameters, respectively. Comparing the SNP locations identified in this study with previous genetic studies, we found that most were mapped proximal to reported QTL and/or genes involved in the salt stress-related responses. The coincidence of the mapped positions supports the hypothesis that the QTL underlying the ST PVE is mostly conserved due to LD and may be under the influence of orthologous genes (Table 5). Interestingly, the PCoA based on the detected SNP markers successfully discriminated the salt-tolerant and salt-sensitive wheat genotypes. The former and latter were mostly distributed in the left and right of the PCoA plot, respectively—an indication that the SNP loci detected in this study are underlying salt stress-responsive candidate genes. One of the promising QTL detected on 2BL chromosome had pleiotropic effects on Fv/Fm, shoot Na+ and CFC in the GWAS analysis. The in silico analysis suggests that the associated QTL interval harbors a putative mixed-linked glucan synthase (CesA) involved in carbohydrate metabolism and mediation of cellulose biosynthesis and cell wall organization (Somerville 2006). Mutation in CesA enhances drought and osmotic stress tolerance in Arabidopsis (Zhu et al. 2010).
GWAS can identify shared mechanisms underlying multiple traits in a systematic way (Pickrell et al. 2016). The integration of genetic studies for multiple traits is considered to be a powerful approach to improve the identification of relevant genetic architectures of complex traits since pleiotropy increases the statistical power to detect genetic variants (Kim et al. 2018). The SNP marker detected at 99.04 cM (PVE = 4.4–14.7%) on 6AL is associated with ABS/RC, DIo/RC, shoot Na+ uptake and shoot K+/Na+ ratio and is proximal to ST QTL reported for Fm, Fv/Fm (Li et al. 2012) and shoot Na+ uptake (Genc et al. 2010). The ORFs harboring this marker is orthologous to the OPAQUE1 gene. OPAQUE1 is salt-responsive and is directly influenced by the vacuolar Na+/H+ antiporter (Sottosanto et al. 2007). The SNP marker (Jagger_c1134_353) at 140.87 cM on 6AL had a genome-wide association with Fv and shoot Na+ uptake and was located in the ORFs of TaABCF3 transporter, suggesting that associated region may be involved in Na+ transport and/or plant ion homeostasis. Reports have indicated that the 6AL harbors three wheat plasma membrane Na+ transporters (Pearce et al. 2014). A gene homolog of TaABCC3 was found in the Q.chl*Qu(2DS) region (at 8.52 cM) on 2DS that had a significant effect on Fm/Fo, Fv/Fo and CFC ST traits in this study. The Q.chl*Qu(2DS) region coincided with the QTL interval previously reported for important agronomic and developmental QTL/genes including QTL Fm (Zhang et al. 2010b), qGY2Da (Zhang et al. 2009), Rht8 and Ppd-D1 (Pestsova and Röder 2002; Gasperini et al. 2012). The TaABCC3 has also been implicated in detoxification, heavy-metal sequestration, chlorophyll catabolite transport, chloroplast catabolite turnover, and ion-channel regulation and has a positive effect on grain formation and mycotoxin tolerance in wheat (Wanke and Üner Kolukisaoglu 2010; Walter et al. 2015). The SNP marker at 68.45 cM on 4BL has a strong genetic effect on shoot K+/Na+ ratio, ABS/RC and TRo/RC. The ORFs sequence carrying this SNP marker is NADH dehydrogenase complex (plastoquinone) assembly (NRAMP-2), and it is involved in photosynthetic electron transport in photosystem I, metal ion transporter/homeostasis and inorganic anion transport (Kriventseva et al. 2014).
Gene-expressional analyses
Salinity stress-induced gene-expressional changes are key components of the molecular mechanisms by which plants adapt to environmental challenges (Hirayama and Shinozaki 2010). The differential gene expression is an essential response of plants against abiotic stresses (Kumari and Pandey 2013). Twenty-eight candidate genes encompassing the SNP markers detected in this study showed differential expressions between salt-tolerant and salt-sensitive wheat genotypes, indicating that the associated regions may be linked to genes conferring salt tolerance and can represent an important resource for the improvement of wheat salt stress adaptation. Among them, OPAQUE1, SPS1, NRAMP-2 and USP were strongly and differentially expressed. Unlike the salt-sensitive genotype, the up-regulation of these genes (relative to control) was observed in the salt-tolerant genotype. Reports have shown that OPAQUE1 exhibits an altered transcript abundance during salinity stress (Cheng et al. 2009; Peng et al. 2009) and acts as a cytoskeleton-cell wall linker to callose synthase complexes in the plasma membrane. The activation and increased expression of OPAQUE1 are parts of plant stress response mechanisms (Kosová et al. 2013). The expressions of OPAQUE1, TaABCF3, and NAD(P)H were also validated with the qRT-PCR approach using two tolerant and two sensitive wheat genotypes (Fig. 4).
The analyses of gene expression kinetics of stress treatments are important and should be considered when expression analyses are performed to identify stress-responsive genes (Deyholos 2010). In this study, the expression kinetics of 15 underlying candidate genes was analyzed at three time points under saline and non-saline conditions. Findings revealed that the gene expressions are genotype, salt stress, and stress duration dependent. Both salt-tolerant and salt-sensitive genotypes exhibited differential expression patterns across the stress durations considered, with salt-sensitive genotypes having higher transcript amount in the first 5 days under salt stress. However, the salt-tolerant genotype displayed greater gene expressions as the salt stress progressed up to 11 days. Three candidate genes including OPAQUE1, hxB, and NRAMP-2 showed an earlier differential response (0–5 days), while MPK9 was differentially expressed after 15 days under salt stress. The differential expression time points observed between the tolerant and sensitive genotypes might coincide with the period described by Munns and Tester (2008) as “ionic phase”—a period when Na+ toxicity (due to high concentrations of Na+ ion) has ensued in the plant tissues, and only plants (salt-tolerant) that are able to exclude the excess Na+ and/or compartmentalize Na+ in vacuoles (as a result of increased activities of stress-responsive genes) can adapt under the salt stress conditions.
The genetic intervals in LD with the pleiotropic SNP markers at 68.45 cM on 4BL and 90.04 cM on 6AL were scanned for other possible causative candidate genes. The expression profiles of all the genes in the associated QTL intervals of scaffold41600 (~ 13 Mb containing 61 candidate genes) on 4BL and scaffold126294.1 (~ 12 Mb containing 141 candidate genes) on 6AL revealed that TraesCS4B01G254300.1 (NRAMP-2) and TraesCS6A01G336500.1 (OPAQUE1) genes showed highest differential expressions between the salt-tolerant and salt-sensitive wheat genotypes after 24 days under salt stress. This result supports the GWAS results and provides strong evidence that the QTL identified on 4BL and 6AL may be harboring salt stress-responsive genes and should be considered for gene-based marker development for salt stress tolerance in wheat; not only because the genes that are linked to the associated pleiotropic markers are showing the highest differential expression between the salt-tolerant and salt-sensitive genotypes but because the biological functions of these genes are related and relevant to the traits evaluated. However, the involvement of both genes in the salt stress response pathways and salt tolerance in wheat need to be validated functionally before exploitation in the development of functional markers for salt stress tolerance.
Analyses of NRAMP-2, TaABCF3 and CSLF1 promoter regions
Transcription factors (TFs) play major roles in the regulation of genes involved in development and tolerance to biotic and abiotic stresses (Dang et al. 2012), by serving as nuclear effectors of multiple signaling cascades and often involved in posttranslational modifications (Tootle and Rebay 2005). The analyses of the promoter are crucial for improving the basic understanding of the expressions of adaptive/stress-responsive genes and will promote their application in plant breeding. Variation in the TF will affect more than one downstream modification as TFs are hierarchically higher and have the ability to regulate many downstream targets (Poser and Bosserhoff 2004), giving rise to gradations of gene expression. In this study, the promoter regions of NRAMP-2, TaABCF3 and CSLF1 were also analyzed for possible detection of ST motifs/cis-regulatory elements in two salt-tolerant and two salt-sensitive wheat genotypes.
The two salt-tolerant genotypes contain unique DNA-binding domains TF_motif_seq_0239/Dof and PBF at 99-bp upstream of the NRAMP-2 translation start codon, whereas these TF features were lacking and/or replaced by the broad-complex 3 and/or hunchback DNA-binding domains in the two salt-sensitive genotypes, indicating that the presence of the Dof-/PBF-motifs may have contributed to the up-regulation of the NRAMP-2 transcriptions in the tolerant genotypes vis-à-vis the sensitive ones. This result provides an initial indication that these motifs may be regulating salt stress response in NRAMP-2, but further validation via reporter gene experiments is needed. Yanagisawa (2000) reported that the Dof proteins are unique to plants and play regulatory roles in the expression of multiple genes involved in pathways for carbon metabolism in maize. The NRAMP-2 gene is involved in the non-photochemical reduction of plastoquinones, cyclic electron transport around photosystem as well as chlororespiration in the thylakoids (Rumeau et al. 2005). In this study, the SNP marker linked to NRAMP-2 had a significant genome-wide association with the specific energy fluxes per reaction center for absorption (ABS/RC) and dissipation-(DIo/RC), and the shoot K+/Na+ ratio, a strong indication that the detected marker could be an eQTL and may serve as a valuable genetic resource for ST improvement in wheat.
The regulatory motif variation in CSLF1 gene was genotype-specific. Genotype UZ-11CWA-08 contains the homeodomain motif family—bZIP, HD-ZIP and GATA-binding proteins (at 581 and 554 bp, respectively) upstream of the translation start codon of the gene, but these motifs were lacking in remaining genotypes. Homeodomain motifs act as growth regulators in response to water deficit, whereas the GATA-binding proteins are associated with light-regulated and tissue-specific response in plants (Teakle et al. 2002; Chow et al. 2015). The linked markers for CSLF1 were found to be genetically associated with Fv/Fm (Maximum quantum yield of PSII). On the other hand, the salt-sensitive genotype UZ-11CWA-24 contains the SPI-B and NF-Y motifs at position 618-bp and 540-bp upstream of the translation start codon, respectively. The report by Ogra et al. (2001) indicated that the SPI motifs regulate the salt stress-dependent posttranscriptional stabilization and have been implicated as a negative regulator of transcription mediated by specific metal-responsive elements. A genome-wide expression analysis has also revealed that the NFY motifs are important plant regulators of abiotic stress tolerance (Petroni et al. 2012; Laloum et al. 2013).
Detection of polymorphisms in the associated gene coding regions
Changes in DNA sequence of the coding genes can cause alterations in the proteins (Studer et al. 2013), and studies aimed at analyzing how these alterations affect protein functions are essential for gaining an in-depth understanding of stress adaptation. The alignment of the expressed sequence tags of OPAQUE1 and NRAMP-2 genes revealed several amino acid substitution sites between Altay2000 and Bobur. For instance, OPAQUE1 (Traes_6AL_891456790.1) had four substitutions: C1529G, A1549V, R1626G and C1628W, based on single nucleotide polymorphisms, at exon 37 that had a significant genome-wide association with multiple ST traits. The R/G substitution is the most non-conservative modified variation, indicating that it may have contributed to the alteration in the gene function during salt stress by replacing the polar positively charged arginine with tiny apolar hydrophobic glycine. However, further studies are very essential to validate the functional consequences of the substitution sites identified in this study in salt stress adaptation.
Author contribution statement
FCO and JL conceived and acquired the project research grant; AB BCO and RCS planned all the research and experiments; BCO designed all the experiments and analyzed/interpreted the data; AB and JL supervised all the experiments; BCO prepared the manuscript; AB, FCO, JL, MB and RCS reviewed the manuscript; and all authors approved the manuscript.
References
Appenroth KJ, Stöckel J, Srivastava A, Strasser RJ (2001) Multiple effects of chromate on the photosynthetic apparatus of Spirodela polyrhiza as probed by OJIP chlorophyll a fluorescence measurements. Environ Pollut 115(1):49–64
Asch F, Dingkuhn M, Dorffling K, Miezan K (2000) Leaf K+/Na+ ratio predicts salinity induced yield loss in irrigated rice. Euphytica 113:109–118
Ashraf MY, Awan AR, Mahmood K (2012) Rehabilitation of saline ecosystems through cultivation of salt tolerant plants. Pak J Bot 44:69–75
Atkinson NJ, Urwin PE (2012) The interaction of plant biotic and abiotic stresses: from genes to the field. J Exp Bot 63:3523–3543
Baxter G, Zhao J, Blanchard C (2011) Salinity alters the protein composition of rice endosperm and the physicochemical properties of rice flour. J Sci Food Agric 91:2292–2297
Berardini TZ, Reiser L, Li D, Mezheritsky Y, Muller R, Strait E, Huala E (2015) The Arabidopsis information resource: making and mining the “gold standard” annotated reference plant genome. Genesis 53(8):474–485
Bradbury PJ et al (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172(2):1165–1177
Burešová I, Sedláčková I, Faměra O, Lipavský J (2010) Effect of growing conditions on starch and protein content in triticale grain and amylose content in starch. Plant Soil Environ 56(3):99–104
Byrt CS, Platten JD, Spielmeyer W, James RA, Lagudah ES, Dennis ES, Tester M, Munns R (2007) HKT1;5-like cation transporters linked to Na+ exclusion loci in wheat, Nax2 and Kna1. Plant Physiol 143(4):1918–1928
Chaerle L, Leinonen I, Jones HG, Van Der Straeten D (2007) Monitoring and screening plant populations with combined thermal and chlorophyll fluorescence imaging. J Exp Bot 58(4):773–784
Chao S, Elias E, Benscher D, Ishikawa G, Huang YF, Saito M et al (2016) Genetic mapping of major-effect seed dormancy quantitative trait loci on chromosome 2B using recombinant substitution lines in tetraploid wheat. Crop Sci 56(1):59–72
Chen TH, Murata N (2011) Glycinebetaine protects plants against abiotic stress: mechanisms and biotechnological applications. Plant Cell Environ 34(1):1–20
Cheng Y, Qi Y, Zhu Q, Chen X, Wang N, Zhao X, Chen H, Cui X, Xu L, Zhang W (2009) New changes in the plasma-membrane-associated proteome of rice roots under salt stress. Proteomics 9:3100–3114
Chow CN, Zheng HQ, Wu NY, Chien CH, Huang HD, Lee TY et al (2015) PlantPAN 2.0: an update of plant promoter analysis navigator for reconstructing transcriptional regulatory networks in plants. Nucleic Acids Res 44(D1):D1154–D1160
Cramer GR (2002) Sodium–calcium interactions under salinity stress. In: Läuchli A, Lüttge U (eds) Salinity: environment-plants-molecules. Kluwer, Dordrecht, pp 205–227
Cuin TA, Shabala S (2006) Potassium homeostasis in salinized plant tissues. In: Volkov A (ed) Plant electrophysiology—theory and methods. Springer, Heidelberg, pp 287–317
Dang PM, Chen CY, Holbrook CC (2012) Identification of drought-induced transcription factors in peanut (Arachis hypogaea L.). J Mol Biochem 1:196–205
Demetriou G, Neonaki C, Navakoudis E, Kotzabasis K (2007) Salt stress impact on the molecular structure and function of the photosynthetic apparatus—the protective role of polyamines. Biochim Biophys Acta (BBA) Bioenerg 1767(4):272–280
Deyholos MK (2010) Making the most of drought and salinity transcriptomics. Plant Cell Environ 33:648–654
FAO (2010) FAO land and plant nutrition management service. www.fao.org. Last Accessed 10 Dec 2010
FAO (2015) FAO Cereal Supply and Demand Brief. http://www.fao.org/worldfoodsituation/csdb/en/. Last Accessed 5 Jan 2016
Farooq M, Bramley H, Palta JA, Siddique KH (2011) Heat stress in wheat during reproductive and grain-filling phases. Crit Rev Plant Sci 30(6):491–507
Flowers TJ (2004) Improving crop salt tolerance. J Exp Bot 55:307–319
Flowers TJ, Galal HK, Bromham L (2010) Evolution of halophytes: multiple origins of salt tolerance in land plants. Funct Plant Biol 37(7):604–612
Fu Y, Frith MC, Haverty PM, Weng Z (2004) MotifViz: an analysis and visualization tool for motif discovery. Nucleic Acids Res 32(2):W420–W423
Fuhrer J, Lehnherr B, Moeri PB, Tschannen W, Shariat-Madari H (1990) Effects of ozone on the grain composition of spring wheat grown in open-top field chambers. Environ Pollut 65(2):181–192
Furbank RT, Tester M (2011) Phenomics—technologies to relieve the phenotyping bottleneck. Trends Plant Sci 16:635–644
Gasperini D, Greenland A, Hedden P, Dreos R, Harwood W, Griffiths S (2012) Genetic and physiological analysis of Rht8 in bread wheat: an alternative source of semi-dwarfism with a reduced sensitivity to brassinosteroids. J Exp Bot 63(12):4419–4436
Genc Y, Oldach K, Verbyla AP, Lott G, Hassan M, Tester M et al (2010) Sodium exclusion QTL associated with improved seedling growth in bread wheat under salinity stress. Theor Appl Genet 121(5):877–894
Glynn CP, Gillian AF, Gavin O (2003) Foliar salt tolerance of Acer genotypes using chlorophyll fluorescence. J Arboricult 29:61–65
Gupta SK, Goyal MR (2017) Soil salinity management in agriculture: technological advances and applications. CRC Press, Boca Raton
Hirayama T, Shinozaki K (2010) Research on plant abiotic stress responses in the post-genome era: past, present and future. Plant J 61(6):1041–1052
Huang S, Spielmeyer W, Lagudah ES, Munns R (2008) Comparative mapping of HKT genes in wheat, barley, and rice, key determinants of Na+ transport, and salt tolerance. J Exp Bot 59(4):927–937
Huang JG et al (2009) GhDREB1 enhances abiotic stress tolerance, delays GA-mediated development and represses cytokinin signalling in transgenic Arabidopsis. Plant Cell Environ 32:1132–1145
Hucl P, Chibbar RN (1996) Variation for starch concentration in spring wheat and its repeatability relative to protein concentration. Cereal Chem 73(6):756–758
Jaggard KW, Qi A, Ober ES (2010) Possible changes to arable crop yields by 2050. Philos Trans R Soc Lond B Biol Sci 365(1554):2835–2851
James RA, Davenport RJ, Munns R (2006) Physiological characterization of two genes for Na+ exclusion in durum wheat, Nax1 and Nax2. Plant Physiol 142:1537–1547
Jighly A, Oyiga BC, Makdis F, Nazari K, Youssef O, Tadesse W et al (2015) Genome-wide DArT and SNP scan for QTL associated with resistance to stripe rust (Puccinia striiformis f. sp. tritici) in elite ICARDA wheat (Triticum aestivum L.) germplasm. Theor Appl Genet 128(7):1277–1295
Kadir S, Von Weihe M, Al-Khatib K (2007) Photochemical efficiency and recovery of photosystem II in grapes after exposure to sudden and gradual heat stress. J Am Soc Hortic Sci 132(6):764–769
Kerfal S, Giraldo P, Rodríguez-Quijano M, Vázquez JF, Adams K, Lukow OM et al (2010) Mapping quantitative trait loci (QTLs) associated with dough quality in a soft × hard bread wheat progeny. J Cereal Sci 52(1):46–52
Kersey PJ, Allen JE, Armean I, Boddu S, Bolt BJ, Carvalho-Silva D et al (2016) Ensembl Genomes 2016: more genomes, more complexity. Nucleic Acids Res 44(D1):D574–D580
Kim HJ, Yu Z, Lawson A, Zhao H, Chung D, Stegle O (2018) Improving SNP prioritization and pleiotropic architecture estimation by incorporating prior knowledge using graph-GPA. Bioinformatics 34(12):2139–2141
Kosová K, Prášil IT, Vítámvás P (2013) Protein contribution to plant salinity response and tolerance acquisition. International journal of molecular sciences 14(4):6757–6789
Kriventseva EV, Tegenfeldt F, Petty TJ, Waterhouse RM, Simao FA, Pozdnyakov IA et al (2014) OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res 43(D1):D250–D256
Kumari A, Pandey DM (2013) Differential gene expression analysis of some least studied transcription factors/regulators in rice during abiotic stresses. Int J Adv Res 1:412–422
Laloum T, De Mita S, Gamas P, Baudin M, Niebel A (2013) CCAAT-box binding transcription factors in plants: Y so many? Trends Plant Sci 18:157–166
Li Y, Lee KK, Walsh S, Smith C, Hadingham S, Sorefan K et al (2006) Establishing glucose-and ABA-regulated transcription networks in Arabidopsis by microarray analysis and promoter classification using a Relevance Vector Machine. Genome Res 16(3):414–427
Li H, Tong Y, Li B, Jing R, Lu C, Li Z (2010) Genetic analysis of tolerance to photo-oxidative stress induced by high light in winter wheat (Triticum aestivum L.). J Genet Genomics 37(6):399–412
Li H, Lin F, Wang G, Jing R, Zheng Q, Li B, Li Z (2012) Quantitative trait loci mapping of dark-induced senescence in winter wheat (Triticum aestivum). J Integr Plant Biol 54(1):33–44
Li Y, Zhang L, Wang X, Zhang W, Hao L, Chu X, Guo X (2013) Cotton GhMPK6a negatively regulates osmotic tolerance and bacterial infection in transgenic Nicotiana benthamiana, and plays a pivotal role in development. FEBS J 280:5128–5144
Li CH, Wang G, Zhao JL, Zhang LQ, Ai LF, Han YF et al (2014a) The receptor-like kinase SIT1 mediates salt sensitivity by activating MAPK3/6 and regulating ethylene homeostasis in rice. Plant Cell 26(6):2538–2553
Li L, Zhang Q, Huang D (2014b) A review of imaging techniques for plant phenotyping. Sensors 14(11):20078–20111
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real time quantitative PCR and the 2−ΔΔCT method. Methods 25(4):402–408
Lobell DB, Bänziger M, Magorokosho C, Vivek B (2011) Nonlinear heat effects on African Maize as evidenced by historical yield trials. Nat Clim Change 1:42–45
Long NV, Dolstra O, Malosetti M, Kilian B, Graner A, Visser RG, van der Linden CG (2013) Association mapping of salt tolerance in barley (Hordeum vulgare L.). Theor Appl Genet 126(9):2335–2351
Lorenz A, Chao S, Asoro F (2011) Chapter two-genomic selection in plant breeding: knowledge and prospects. Adv Agron 110:77–123
Lutts S, Kinet JM, Bouharmont J (1995) Changes in plant response to NaCl during development of rice (Oryza sativa L.) varieties differing in salinity resistance. J Exp Bot 46:1843–1852
Maathuis FJM, Amtmann A (1999) K+ nutrition and Na+ toxicity: the basis of cellular K+/Na+ ratios. Ann Bot (Lond) 84:123–133
Marschner P, Marschner’s (2012) Mineral nutrition of higher plants, 3rd edn. Academic Press, London, pp 178–189
Masoudi B, Mardi M, Hervan EM, Bihamta MR, Naghavi MR, Nakhoda B, Amini A (2015) QTL mapping of salt tolerance traits with different effects at the seedling stage of bread wheat. Plant Mol Biol Rep 33(6):1790–1803
Mathur S, Jajoo A (2014) Alterations in photochemical efficiency of photosystem II in wheat plant on hot summer day. Physiol Mol Biol Plants 20(4):527–531
Mathur S, Mehta P, Jajoo A (2013) Effects of dual stress (high salt and high temperature) on the photochemical efficiency of wheat leaves (Triticum aestivum). Physiol Mol Biol Plants 19(2):179–188
McCouch S, Baute GJ, Bradeen J, Bramel P, Bretting PK, Buckler E, Burke JM, Charest D, Cloutier S, Cole G et al (2013) Agriculture: feeding the future. Nature 499:23–24
McCouch SR, Wright MH, Tung CW, Maron LG, McNally KL, Fitzgerald M et al (2016) Open access resources for genome-wide association mapping in rice. Nat Commun 7:10532
Mehta P, Kraslavsky V, Bharti S, Allakhverdiev SI, Jajoo A (2011) Analysis of salt stress-induced changes in photosystem II heterogeneity by prompt fluorescence and delayed fluorescence in wheat (Triticum aestivum) leaves. J Photochem Photobiol B 104(1):308–313
Munns R, Tester M (2008) Mechanisms of salinity tolerance. Annu Rev Plant Biol 59:651–681
Nayidu N, Bollina V, Kagale S (2013) Oilseed crop productivity under salt stress. In: Ecophysiology and responses of plants under salt stress. Springer, New York, pp 249–265
O’Neill M (2010) ANOVA and REML: a guide to linear mixed models in an experimental design context, statistical advisory and training service Pty Ltd, 180 pp
Ogra Y, Suzuki K, Gong P, Otsuka F, Koizumi S (2001) Negative regulatory role of Sp1 in metal responsive element-mediated transcriptional activation. J Biol Chem 276(19):16534–16539
Okamoto Y, Kajimura T, Ikeda TM, Takumi S (2012) Evidence from principal component analysis for improvement of grain shape- and spikelet morphology-related traits after hexaploid wheat speciation. Genes Genet Syst 87:299–310
Oyiga BC, Sharma RC, Shen J, Baum M, Ogbonnaya FC, Léon J, Ballvora A (2016) Identification and characterization of salt tolerance of wheat germplasm using a multivariable screening approach. J Agron Crop Sci 202(6):472–485
Oyiga BC, Sharma RC, Baum M, Ogbonnaya FC, Léon J, Ballvora A (2018) Allelic variations and differential expressions detected at quantitative trait loci for salt stress tolerance in wheat. Plant Cell Environ 41(5):919–935
Parida AK, Das AB (2005) Salt tolerance and salinity effects on plants: a review. Ecotoxicol Environ Saf 60:324–349
Pasam RK, Sharma R (2014) Association mapping: a new paradigm for dissection of complex traits in crops. In: Agricultural bioinformatics. Springer India, pp 1–20
Pauli D, Chapman SC, Bart R, Topp CN, Lawrence-Dill CJ, Poland J, Gore MA (2016) The quest for understanding phenotypic variation via integrated approaches in the field environment. Plant Physiol 172(2):622–634
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Pearce S, Tabbita F, Cantu D, Buffalo V, Avni R, Vazquez-Gross H et al (2014) Regulation of Zn and Fe transporters by the GPC1 gene during early wheat monocarpic senescence. BMC Plant Biol 14(1):368
Peng Z, Wang M, Li F, Lv H, Li C, Xia G (2009) A proteomic study of the response to salinity and drought stress in an lntrogression strain of bread wheat. Mol Cell Proteomics 8:2676–2686
Pestsova E, Röder M (2002) Microsatellite analysis of wheat chromosome 2D allows the reconstruction of chromosomal inheritance in pedigrees of breeding programmes. Theor Appl Genet 106(1):84–91
Petroni K, Kumimoto RW, Gnesutta N, Calvenzani V, Fornari M, Tonelli C, Holt BF, Mantovani R (2012) The promiscuous life of plant NUCLEAR FACTOR Y transcription factors. Plant Cell 24:4777–4792
Pickrell JK, Berisa T, Liu JZ, Ségurel L, Tung JY, Hinds D (2016) Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet 48(7):709
Piepho H, Mohring J, Melchinger A, Buchse A (2008) BLUP for phenotypic selection in plant breeding and variety testing. Euphytica 161:209–228
Piikki K et al (2008) The grain quality of spring wheat (Triticum aestivum L.) in relation to elevated ozone uptake and carbon dioxide exposure. Eur J Agron 28(3):245–254
Poser I, Bosserhoff AK (2004) Transcription factors involved in development and progression of malignant melanoma. Histol Histopathol 19:173–188
Prasad M, Kumar N, Kulwal PL, Roder MS, Balyan HS, Dhaliwal HS, Gupta PK (2003) QTL analysis for grain protein content using SSR markers and validation studies using NILs in bread wheat. Theor Appl Genet 106:659–667
Qadir M, Tubeileh A, Akhtar J, Larbi A, Minhas PS, Khan MA (2008) Productivity enhancement of salt-affected environments through crop diversification. Land Degrad Dev 19(4):429–453
Rahaman MM, Chen DJ, Gillani Z, Klukas C, Chen M (2015) Advanced phenotyping and phenotype data analysis for the study of plant growth and development. Front Plant Sci 6:619
Rao PS, Mishra B, Gupta SR (2013) Effects of soil salinity and alkalinity on grain quality of tolerant, semi-tolerant and sensitive rice genotypes. Rice Sci 20(4):284–291
Rumeau D, Bécuwe-Linka N, Beyly A, Louwagie M, Garin J, Peltier G (2005) New subunits NDH-M,-N, and-O, encoded by nuclear genes, are essential for plastid Ndh complex functioning in higher plants. Plant Cell 17(1):219–232
Sandhu N, Jain S, Kumar A, Mehla BS, Jain R (2013) Genetic variation, linkage mapping of QTL and correlation studies for yield, root, and agronomic traits for aerobic adaptation. BMC Genet 14(1):104
Satoh K, Smith CM, Fork DC (1983) Effects of salinity on primary processes of photosynthesis in the red alga Porphyra perforata. Plant Physiol 73(3):643–647
Shi W et al (2014) The Gossypium hirsutum WRKY gene GhWRKY39-1 promotes pathogen infection defense responses and mediates salt stress tolerance in transgenic Nicotiana benthamiana. Plant Cell Rep 33:483–498
Shu S, Guo SR, Sun J, Yuan LY (2012) Effects of salt stress on the structure and function of the photosynthetic apparatus in Cucumis sativus and its protection by exogenous putrescine. Physiol Plant 146:285–296
Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH (2009) JBrowse: a next-generation genome browser. Genome Res 19(9):1630–1638
Somerville C (2006) Cellulose synthesis in higher plants. Annu Rev Cell Dev Biol 22:53–78
Sottosanto J, Saranga Y, Blumwald E (2007) Impact of Atnhx1, A vacuolar Na+/H+ antiporter, upon gene expression during short- and long-term salt stress in Arabidopsis Thaliana. BMC Plant Biol 7:18–25
Stothard P (2000) The Sequence Manipulation Suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28:1102–1104
Strasser RJ, Srivastava A, Tsimilli-Michael M (2004) Analysis of the chlorophyll a fluorescence transient. In: Papageorgiou G, Govindjee (eds) Chlorophyll fluorescence a signature of photosynthesis. Advances in photosynthesis and respiration. Springer, Dordrecht, pp 321–362
Studer RA, Dessailly BH, Orengo CA (2013) Residue mutations and their impact on protein structure and function: detecting beneficial and pathogenic changes. Biochem J 449(3):581–594
Sun H, Lü J, Fan Y, Zhao Y, Kong F, Li R et al (2008) Quantitative trait loci (QTLs) for quality traits related to protein and starch in wheat. Prog Nat Sci 18(7):825–831
Sun C, Zhang F, Yan X, Zhang X, Dong Z, Cui D, Chen F (2017) Genome-wide association study for 13 agronomic traits reveals distribution of superior alleles in bread wheat from the Yellow and Huai Valley of China. Plant Biotechnol J 15(8):953–969
Tan MK, Sharp PJ, Lu MQ, Howes N (2006) Genetics of grain dormancy in a white wheat. Crop Pasture Sci 57(11):1157–1165
Teakle GR, Manfield IW, Graham JF, Gilmartin PM (2002) Arabidopsis thaliana GATA factors: organization, expression and DNA-binding characteristics. Plant Mol Biol 50(1):43–56
Thitisaksakul M, Tananuwong K, Shoemaker CF, Chun A, Tanadul OUM, Labavitch JM, Beckles DM (2015) Effects of timing and severity of salinity stress on rice (Oryza sativa L.) yield, grain composition, and starch functionality. J Agric Food Chem 63(8):2296–2304
Tootle TL, Rebay I (2005) Post-translational modifications influence transcription factor activity: a view from the ETS superfamily. BioEssays 27:285–298
Tsilo TJ, Simsek S, Ohm JB, Hareland GA, Chao S, Anderson JA (2011) Quantitative trait loci influencing endosperm texture, dough-mixing strength, and bread-making properties of the hard red spring wheat breeding lines. Genome 54(6):460–470
Turki N, Shehzad T, Harrabi M, Okuno K (2015) Detection of QTLs associated with salinity tolerance in durum wheat based on association analysis. Euphytica 201(1):29–41
Volkov V, Amtmann A (2006) Thellungiella halophila, a salt-tolerant relative of Arabidopsis thaliana, has specific root ion-channel features supporting K+/Na+ homeostasis under salinity stress. Plant J 48:342–353
von Korff M, Radovic S, Choumane W, Stamati K, Udupa SM, Grando S, Ceccarelli S, Mackay I, Powell W, Baum M, Morgante M (2009) Asymmetric allele-specific expression in relation to developmental variation and drought stress in barley hybrids. Plant J 59:14–26
Walter S, Kahla A, Arunachalam C, Perochon A, Khan MR, Scofield SR, Doohan FM (2015) A wheat ABC transporter contributes to both grain formation and mycotoxin tolerance. J Exp Bot 66(9):2583–2593
Wanke D, Üner Kolukisaoglu H (2010) An update on the ABCC transporter family in plants: many genes, many proteins, but how many functions? Plant Biol 12:15–25
Payne R, Welham, S, Harding S (2015) A guide to REML in Genstat® for WindowsTM 18th edition. VSN International, Hemel Hempstead, UK. www.vsni.co.uk/software/genstat
Xu Y, Li S, Li L, Zhang X, Xu H, An D (2013) Mapping QTLs for salt tolerance with additive, epistatic and QTL × treatment interaction effects at seedling stage in wheat. Plant Breeding 132(3):276–283
Yamane K, Kawasaki M, Taniguchi M, Miyake H (2008) Correlation between chloroplast ultrastructure and chlorophyll fluorescence characteristics in the leaves of rice (Oryza sativa L.) grown under salinity. Plant Prod Sci 11:139–145
Yanagisawa S (2000) Dof1 and Dof2 transcription factors are associated with expression of multiple genes involved in carbon metabolism in maize. Plant J 21(3):281–288
Zhang K, Zhang Y, Chen G, Tian J (2009) Genetic analysis of grain yield and leaf chlorophyll content in common wheat. Cereal Res Commun 37(4):499–511
Zhang JL, Flowers TJ, Wang SM (2010a) Mechanisms of sodium uptake by roots of higher plants. Plant Soil 326(1–2):45
Zhang ZB, Xu P, Jia JZ, Zhou RH (2010b) Quantitative trait loci for leaf chlorophyll fluorescence traits in wheat. Aust J Crop Sci 4(8):571
Zhang L, Li Y, Lu W, Meng F, Wu CA, Guo X (2012) Cotton GhMKK5 affects disease resistance, induces HR-like cell death, and reduces the tolerance to salt and drought stress in transgenic Nicotiana benthamiana. J Exp Bot 63:3935–3951
Zheng F, Wang X, Zhang W, Hou P, Lu F, Du K, Sun Z (2013) Effects of elevated O3 exposure on nutrient elements and quality of winter wheat and rice grain in Yangtze River Delta, China. Environ Pollut 179:19–26
Zhu J, Lee BH, Dellinger M, Cui X, Zhang C, Wu S et al (2010) A cellulose synthase-like protein is required for osmotic stress tolerance in Arabidopsis. Plant J 63(1):128–140
Acknowledgements
We acknowledge support from Alex Morgounov, Mesut Keser and Wuletaw Tadesse in providing germplasm for this study. We greatly appreciate the support of collaborators in Karshi (Zokhidjon Ziyadullaev) and Urgench (Yulduzoy Djumaniyazova and Jumanazar Ruzimov) in Uzbekistan in conducting the salinity field trials. A large proportion of germplasm used in the study originated from the International Winter Wheat Improvement Program (a joint breeding program of Ministry of Agriculture of Turkey, CIMMYT, and ICARDA). We also acknowledge Dr Greg Rebetzke (CSIRO Agriculture and Food) for critical review of this manuscript. The authors are grateful to the “Bundesministerium für wirtschaftliche Zusammenarbeit und Entwicklung” (BMZ) via “German Agency for International Cooperation” (GIZ), Germany, for financing this research (Project Number: 09.7860.1-001.00). Funding for open access publication was provided by ICARDA within the framework of “Collaborative Research Project on Sustainable Soil Management to Enhance Agricultural Productivity in Central Asia, Phase II” in partnership with the International Food Policy Research (IFPRI) and Eurasian Center for Food Security under funding support from Russian Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Communicated by Diane E. Mather.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Oyiga, B.C., Ogbonnaya, F.C., Sharma, R.C. et al. Genetic and transcriptional variations in NRAMP-2 and OPAQUE1 genes are associated with salt stress response in wheat. Theor Appl Genet 132, 323–346 (2019). https://doi.org/10.1007/s00122-018-3220-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-018-3220-5