
Abstract
Viroporins are short polypeptides encoded by viruses. These small membrane proteins assemble into oligomers that can permeabilize cellular lipid bilayers, disrupting the physiology of the host to the advantage of the virus. Consequently, efforts during the last few decades have been focused towards the discovery of viroporin channel inhibitors, but in general these have not been successful to produce licensed drugs. Viroporins are also involved in viral pathogenesis by engaging in critical interactions with viral proteins, or disrupting normal host cellular pathways through coordinated interactions with host proteins. These protein-protein interactions (PPIs) may become alternative attractive drug targets for the development of antivirals. In this sense, while thus far most antiviral molecules have targeted viral proteins, focus is moving towards targeting host proteins that are essential for virus replication. In principle, this largely would overcome the problem of resistance, with the possibility of using repositioned existing drugs. The precise role of these PPIs, their strain- and host- specificities, and the structural determination of the complexes involved, are areas that will keep the fields of virology and structural biology occupied for years to come. In the present review, we provide an update of the efforts in the characterization of the main PPIs for most viroporins, as well as the role of viroporins in these PPIs interactions.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Viroporins
- Interactome
- Cellular pathways
- Genome-wide screens
- Membrane protein structure
- Protein-protein interactions
The Viroporins and Channel Activity Inhibition
The field of viroporin research has its origins in the observation that virus-infected cells show increased membrane permeability (Carrasco 1995). More than two decades later, viroporins have been confirmed in several viral families, e.g., Orthomyxoviridae (AM2, PB1-F2, BM2), Flaviviridae (p7), Coronaviridae (E, 3a, 4a), Paramyxoviridae (SH), Picornaviridae (2B/2 BC, 3A), Togaviridae (6 K), Retroviridae (Vpr, Vpu, p13), Reoviridae (NSP4 and p10), Polyomaviridae (agnoprotein, VP2-VP4), Papillomaviridae (E5), or Rhabdoviridae (α1). Currently, detailed structural information is limited to only a handful of viroporins (vide infra), although these constitute useful templates for the probably hundreds of other unknown viroporins yet to be discovered in reservoir hosts (Anthony et al. 2013).
In most cases, viral attenuation is not only achieved by deletion of the viroporin gene but also simply when their channel activity is suppressed. Indeed, various specific pathogenic roles of viroporin channel activity have been discovered, and attempts have been made to modulate this channel activity, especially that of influenza A virus M2 (IAV M2, or AM2) protein, the first discovered and the best characterized viroporin. In general, however, the road to rational design and discovery of viroporin small-molecule inhibitors has not been successful [see To et al. (2016) for a recent review]. In fact, amantadine and rimantadine are at present the only licensed antiviral drugs that target a viroporin, i.e., IAV M2. However, most circulating strains of IAV are Amtresistant (Deyde et al. 2007; Hayden and De Jong 2011), and neither drug is currently being used in humans.
Protein-Protein Interactions (PPIs) Involving Viroporins
Viroporins are involved in many protein-protein interactions (PPIs) that may be also susceptible to therapeutic intervention [see recent reviews (Fischer et al. 2014; Nieva and Carrasco 2015)]. Both intraviral and virus-host interactions, in the form of a myriad of perturbations, can provide important insights into the mechanisms involved in the viral infectious cycle. Also, understanding these PPI networks may aid the design of new antivirals. These strategies depend on both detailed structural and mechanistic information and on the availability of therapeutically relevant targets. In this sense, the initial focused approach to identify viroporin binders is being complemented with genome-wide interactome studies adapted to viral infections using high-throughput technologies, providing a dramatic boost in the search for possible PPIs [see de Chassey et al. (2014) for a recent review].
Useful methods to obtain leads for PPIs involving viroporins include yeast two-hybrid (Y2H) screens, e.g., the genome-wide virus-host PPI screen of HCV was performed almost 10 years ago using a construction of a viral ORFeome and Y2H technology (De Chassey et al. 2008), identifying hundreds of PPIs involving viral and host proteins, 13 of which involving p7 and proteins expressed in the liver. Other Y2H screens have included IAV M2 virus-virus and virus-host interactions (Shapira et al. 2009), or intraviral PPIs in the coronavirus responsible for severe acute respiratory syndrome (SARS-CoV) (von Brunn et al. 2007). However, this method does not measure interactions between proteins in the context of the infected cell, is biased against membrane proteins, and cannot study protein complexes that are weakly or transiently associated. The bias against membrane proteins can be compensated using the split-ubiquitin-based yeast two-hybrid screen, e.g., in a screen to search for binders of the small hydrophobic (SH) protein of the respiratory syncytial virus (RSV) (Li et al. 2015).
More suitable methods detect interactions in the context of the infected cell, using a combination of affinity purification with mass spectrometry, e.g., Wang et al. (2017), although the method has also low sensitivity. Other methods are based on microarrays of deposited purified proteins (Zhu et al. 2001) and protein complementation assay (PCA) (Tarassov et al. 2008). Lastly, an approach that combines on-chip in vitro protein synthesis with an in situ microfluidic affinity assay can detect even weak or transient interactions (Gerber et al. 2009), although host-virus PPIs using this method so far has only been tested for M protein in RSV (Kipper et al. 2015).
From these studies, it is apparent that viroporins, and viral proteins in general, tend to show preference for key host proteins that have a high number of direct interacting partners. Also, interactions may be simultaneous with many cellular proteins, making use of intrinsically disordered protein regions enriched for short linear motifs, e.g., PDZ-binding motifs (Hagai et al. 2014; Meyniel-Schicklin et al. 2012), to compensate for their small proteomes.
The Influenza A Virus Matrix Protein 2 (IAV M2 or AM2)
Influenza viruses belong to the Orthomyxoviridae family of segmented, negative-sense, enveloped RNA viruses. The seasonal flu caused by the influenza A virus (IAV) is known for causing pandemics with high mortality rates (Hay et al. 2001; Neumann et al. 2009), although its close relative influenza B accounts for half of the influenza disease in recent years (www.cdc.gov). Generally, influenza virions are spherical in shape ranging from 80 to 120 nm in diameter, although filamentous forms may also occur (Lamb and Choppin 1983). The viral envelope contains three transmembrane proteins, hemagglutinin (HA), neuraminidase (NA), and matrix protein 2 (M2), on the outside and a layer of matrix protein (M1) just underneath the membrane that contains cholesterol-enriched lipid rafts. M1 forms an internal coat that encloses the viral ribonucleoproteins (vRNPs), i.e., the negative-strand viral RNA (vRNA) and nucleoprotein (NP), with small amounts of the nuclear export protein (NEP) and three polymerase (3P) proteins (PA, PB1, and PB2) that form the viral RNA polymerase complex (Fields et al. 2013).
M2 Viroporin
The viroporin M2 in IAV is a homotetrameric channel (Sakaguchi et al. 1997). Each M2 monomer is a 97-amino acid protein comprising an N-terminal ectodomain (24 aa), an α-helical transmembrane domain (TMD, 19 aa), and a highly conserved cytoplasmic tail (CT) domain (54 aa) that is a hotspot for interactions with both viral and host proteins during the IAV life cycle. The latter may therefore constitute an attractive drug target for the development of IAV antivirals. M2 has a pH-activated proton channel activity which is required to complete the uncoating process during virus entry. Upon virus internalization via endocytosis, M2 selectively conducts protons from acidified endosomes into the viral interior. This acidification of virion triggers the dissociation of the M1 protein from the vRNP complex, thereby enabling the transport of vRNPs into the nucleus for replication of viral genetic material (Helenius 1992). For some IAV subtypes, the M2 proton channel raises the pH of the trans-Golgi network (TGN) to protect the viral HA from premature low-pH conformational change during its transport to the cell surface (Takeuchi and Lamb 1994). The channel activity of IAV M2 has been found to be sufficient for the activation of the NLRP3 inflammasome in influenza-infected cells (Ichinohe et al. 2010). Presently, there are more than ten structures of both wild-type and drug-resistant mutant M2 channels in the Protein Data Bank [see review in Gu et al. (2013)].
M1-M2 Interaction
Early works suggested a role for IAV M1-M2 interaction in virus budding and control of virion morphology (filamentous versus spherical). Interaction between M1 with the M2 cytoplasmic tail was first suggested from the analysis of escape mutants (Zebedee and Lamb 1989). Further work revealed a physical interaction between M1 and the M2 cytoplasmic tail at the site of virus budding, to facilitate virus assembly by promoting the recruitment and packaging of viral proteins and viral genome (McCown and Pekosz 2006; Chen et al. 2008). The cytoplasmic tail of M2 contains an amphipathic α-helix (residues 45–62) (Schnell and Chou 2008) that can modulate membrane curvature in a cholesterol-dependent manner. This feature of M2 has been proposed to be implicated in (i) modification of local membrane curvature during virus budding to provide a stabilized scaffold for M1 polymerization and virus filament formation and (ii) alteration of membrane curvature at the neck of budding virions to facilitate membrane scission and virion release (Rossman and Lamb 2011).
Host Interactions
In addition to intraviral interactions, a number of interactions of M2 with host proteins have been described to modulate autophagy, membrane trafficking, host defense, and virus budding. For example, IAV M2 has been reported to arrest autophagy (Gannagé et al. 2009; Beale et al. 2014), a cellular degradation pathway mediated by autophagosomes which delivers cytoplasmic materials to the lysosome that is regulated by autophagy-related genes (Atg). This process involves (i) target engulfment by an isolation crescent membrane (phagophore) to form the autophagosome and (ii) autophagosome-lysosome fusion and degradation of the intra-autophagosomal contents (Fig. 15.1a). IAV subverts this machinery by blocking this fusion, resulting in increased apoptosis of IAV-infected cells. In IAV-infected A549 human lung epithelial cells, M2 coimmunoprecipitates with Atg6/Beclin-1 through interaction with M2 residues 1–60 (Gannagé et al. 2009). Atg6/Beclin1 is part of a complex that regulates autophagosome generation and degradation and is a common target of other viruses for the subversion of autophagy, e.g., herpesviruses and the human immunodeficiency virus (HIV) [reviewed in Münz (2011)].

The C-terminal tail of M2 has also been implicated in the binding to LC3, a protein that normally localizes to autophagosomal membranes (Sou et al. 2006) to recruit autophagy receptors carrying substrates destined for autophagic degradation. These receptors typically contain an LC3-interacting region (LIR), with consensus LIR motif W/FxxI/L/V. In IAV-infected cells, the localization of LC3 changes from the cytoplasm and autophagosomal membranes to the plasma membrane (Beale et al. 2014), a change mediated by a putative LIR motif (residues 91–94, FVSI) present in the cytoplasmic tail of M2 (Fig. 15.1b). Binding of LC3 to the LIR motif of M2 has been confirmed by LUMIER binding assays (Barrios-Rodiles et al. 2005) and GFP pull-down experiments.
The M2-LC3 interaction is also a factor in the budding of IAV, which, depending on the viral strain and host cell type, can produce either spheres or filaments (Bourmakina and García-Sastre 2003), with the latter requiring extensive membrane resources. Cells infected with a filamentous budding IAV strain carrying mutations in the M2 LIR motif that abolished M2-LC3 interactions produced fewer filaments than cells infected with wild-type IAV, suggesting that hijacking of LC3 by M2 may assist in the delivery of LC3-conjugated membranes to the cell surface to facilitate IAV budding (Beale et al. 2014).
The cytoplasmic domain of IAV M2 has been reported to bind caveolin-1 (Cav-1) (Zou et al. 2009), a raft-residing cholesterol-binding protein implicated in the life cycle of viruses that buds from lipid rafts, such as HIV, RSV, and rotavirus. Most Cav-1-associated proteins contain an aromatic-rich caveolin-binding motif (CBM), a consensus sequence of aromatic residues separated by a specific spacing (Couet et al. 1997). The M2-Cav-1 interaction has been confirmed by pull-down and coimmunoprecipitation assays, with the putative CBM in M2 proposed to reside in the cytoplasmic, juxtamembrane region of the M2 tail (Sun et al. 2010). This interaction suggests that Cav-1 may modulate virus budding, possibly through the trafficking of M2 to the plasma membrane.
A yeast two-hybrid screening effort identified the transport protein particle complex 6A (TRAPPC6A) and also its N-internal deleted isoform, TRAPPC6AΔ, as binders to the last six amino acids at the C-terminal end of IAV M2, with highly conserved Leu96 located at the extremity of M2 being indispensable in mediating the interaction (Zhu et al. 2017). TRAPP complexes are multi-subunit tethering complexes involved in intracellular membrane trafficking pathways, and TRAPPC6AΔ may be a regulator of M2 transport to the cell surface.
A yeast two-hybrid assay combined with mutagenesis identified the binding of both AM2 and BM2 to the C-terminal domain 1 (CTD1) of Hsp40 (Guan et al. 2010), a molecular chaperone that can regulate the critical PKR signaling pathway against viral infection. Hsp40 associates with the PKR inhibitor, P58IPK. Binding studies suggest that M2 also binds to P58IPK, possibly forming a stable complex with both Hsp40 and P58IPK (Guan et al. 2010), which would enhance PKR autophosphorylation and activation to inhibit host protein synthesis.
Results from another yeast two-hybrid screen using the M2 cytoplasmic tail as a bait have identified the human cell cycle regulator cyclin D3 as a binder, and their interaction has been confirmed in infected cells by immunoprecipitation assays (Fan et al. 2017). Using siRNA-mediated knockdown of cyclin D3 expression, the study proposed that cyclin D3 is a negative modulator of IAV infection, competing with M1 for binding to M2 in transfected cells and therefore disrupting the important M1-M2 interaction in the context of an IAV infection. On the other hand, IAV may antagonize cyclin D3 activity by (i) relocating cyclin D3 from the nucleus (Fig. 15.2a) to the cytosol (Fig. 15.2b) to facilitate virus replication by promoting cell cycle arrest and (ii) targeting cyclin D3 for cytosolic proteasomal degradation to prevent its interference with M1-M2 binding (Fig. 15.2b). Cyclin D3 has been also reported to be a target of SARS-CoV viroporin 3a (Yuan et al. 2007)

Proposed activity of cyclin D3 in IAV budding. (a) In normal cells, cyclin D3 is localized to the nucleus where it regulates cell cycle; (b) during IAV infection, cyclin D3 interacts with viral M2 to disrupt M1-M2 binding; (c) in the absence of cyclin D3, more infectious progeny virus particles are released [adapted from Fan et al. (2017)]
A recent study has identified M2 as a putative viral antagonist of BST-2 (bone marrow stromal cell antigen 2, also known as tetherin, CD317) (Hu et al. 2017), a protein that may restrict the release of infectious IAV (Mangeat et al. 2012). BST-2 can inhibit the release of a wide range of enveloped viruses by tethering budding virions to the cell surface [reviewed in le Tortorec et al. (2011)], e.g., in HIV-1, in that case antagonized by its viroporin Vpu (Neil et al. 2008). However, the role of BST-2 in limiting IAV release has been disputed (Watanabe et al. 2011), perhaps due to IAV strain-specific susceptibility to BST-2 restriction. This interaction was confirmed by orthogonal assays, and using chimeric and truncated M2, the regions involved in BST-2 downregulation were proposed to be within the M2 extracellular and TMDs.
Another report, using a Y2H screening with M2 cytoplasmic tail as bait, discovered the binding to annexin A6 (AnxA6) (Ma et al. 2012), a Ca2+-regulated membrane-binding protein that controls intracellular cholesterol homeostasis, and regulates membrane fusion and vesicle formation in endocytic and exocytic pathways (Raynal and Pollard 1994). This interaction has been verified by coimmunoprecipitation assays and colocalization studies in infected cells (Ma et al. 2012). Modulation of AnxA6 expression led to corresponding variations in production of infectious IAV, suggesting that AnxA6 is a negative modulator of IAV infection (Ma et al. 2012), as it may impair IAV budding and the release of progeny virus.
A Y2H screen also identified Na+/K+ ATPase β1 subunit (ATP1B1) as binder to the M2 cytoplasmic domain (Mi et al. 2010). SARS-CoV E viroporin and the human papillomavirus E5 viroporin have also been found to bind the host Na+/K+ ATPase α1 subunit (Nieto-Torres et al. 2011) and vacuolar H+ ATPase (Andresson et al. 1995), respectively.
PB1-F2 Viroporin
Another protein in IAV, PB1-F2 (Chen et al. 2001), has the hallmarks of a viroporin: it is ~90 residues long, it forms oligomers, and it has been shown to form a nonselective ion channel in planar lipid bilayers and microsomes (Henkel et al. 2010). PB1-F2 is known to induce apoptosis in host immune cells (Chen et al. 2001) via interaction with two mitochondrial proteins, ANT3 and VDAC1, resulting in the loss of mitochondria membrane potential (Varga et al. 2012), although its localization and pro-apoptotic behavior is strain and cell type specific (Varga and Palese 2011). In addition, PB1-F2 from pathogenic strains of IAV can be incorporated into the phagolysosomal compartment to activate the NLRP3 inflammasome, resulting in IL-1β secretion and causing severe pathophysiology (McAuley et al. 2013). Also, binding of PB1-F2 of PR8 to MAVS, a RIG-I-like receptor (RLR) signaling adaptor anchoring to mitochondria, led to antagonism activity on interferon production (Varga et al. 2012). Despite these data, the precise role of PB1-F2 in modulation of IAV-induced immunopathogenesis is still unknown.
IAV Protein Interactome
Many more IAV host-virus PPIs have been detected recently using affinity purification coupled with mass spectrometry in the context of infected cells. For example, the interactome of 11 viral proteins of influenza PR8 IAV and another 3 strains was analyzed (Wang et al. 2017), confirming that M2 protein is one of the major nodes connecting host proteins with roles in immunity and regulation of viral infection. Almost 100 interactions of host with M2 protein were detected, and ~30 were common to at least three of the strains (Fig. 15.3).

Maps of the IAV intraviral and virus-host protein interactome. (a) Result from a Y2H study to identify direct binary contacts among the ten major viral proteins of the PR8 strain (Shapira et al. 2009). This study also detected nine interactions between IAV M2 and host proteins, among them RNA-binding proteins, transcription factors, or proteins involved in signaling pathways, or to detect intraviral PPIs; (b) close-up of high-confidence candidate-interacting proteins (HCIPs) associated with multiple IAV strains when M2 was used as bait. Indicated are links to other viral proteins (NP, PB1, and PB2) [adapted from Wang et al. (2017)]
The Hepatitis C Virus p7 Protein (HCV-p7)
The hepatitis C virus (HCV) is an enveloped positive-strand RNA virus member of the Flaviviridae family (genus Hepacivirus) that has chronically infected 170 million people worldwide, causing human liver disease. Hepatitis C is divided into six genotypes, with genotype 1 being the most common and most difficult to treat. Treatment against HCV infection involves drugs targeting both viral and host proteins, e.g., drugs that target NS3/4A protease, the NS5A protein, or the NS5B RNA-dependent RNA polymerase (RdRP). However, the rapid turnover of HCV replication (Neumann et al. 1998) and the error-prone activity of the HCV RNA polymerase lead to a rapid formation of “quasi-species” and therefore resistance to antivirals.
HCV encodes a single polyprotein of ~3000 amino acids that is cleaved by cellular and viral proteases into ten different proteins: three structural proteins (core, E1, and E2) and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). The structural proteins Core (C), E1, and E2 are located in the N-terminal region and form the viral particle (Moradpour and Penin 2013), whereas NS3 to NS5B are involved in the replication of the viral genome. p7 and NS2 are dispensable for RNA replication but are critical for virion morphogenesis, which requires both structural and nonstructural proteins (Appel et al. 2008; Steinmann et al. 2007), although the latter are not packaged in viral particles.
HCV replication takes place in double-membrane vesicles (DMVs), while viral assembly sites (AS) have been suggested to be specialized detergent-resistant membranes (DRMs) in the ER or in mitochondria-associated ER membranes rich in cholesterol and sphingolipids (Shanmugam et al. 2015). The core protein concentrates at cytosolic lipid droplets (cLDs) close to the ER-located assembly site and is eventually linked to the vRNA replication site in specialized ER-derived structures.
During HCV assembly, one of the first steps is the interaction of cLD-bound core protein and NS5A (Appel et al. 2008). NS2, probably in complex with p7, interacts with the NS3-4A enzyme, and this retrieves the viral core protein from cLDs into the nascent virus particles [reviewed in Lindenbach and Rice (2013)]. Virus particles transit through the secretory pathway, where they are protected from exposure to low pH by p7, which neutralizes intracellular compartments (Wozniak et al. 2010). More recently, a genetic interaction has been observed between p7 and NS5B proteins, which were found to cooperate to promote virion infectivity by decreasing sphingomyelin content in the virion (Aligeti et al. 2015).
p7 Viroporin
The viroporin p7 is produced when E2-p7-NS2 is cleaved by a signal peptidase at the ER (Lin et al. 1994; Mizushima et al. 1994). p7 is a 63-residue-long protein that has two α-helical TMDs and is found mainly at the ER membrane. As mentioned above, p7 is essential for virus particle assembly and release (Steinmann et al. 2007) and for productive HCV propagation in vivo (Sakai et al. 2003), but not necessary for RNA replication. The bovine viral diarrhea virus (BVDV) and the hepacivirus GB virus B (GBV-B), HCV’s closest relatives, also have a p7 protein crucial for virus replication.
p7 has channel activity with low cation selectivity (Griffin et al. 2003; Ouyang et al. 2013). p7 has been reported to permeabilize membranes to protons, preventing the acidification of intracellular vesicles (Wozniak et al. 2010), an activity that has been confirmed in vitro using a liposome-based assay (Gan et al. 2014).
The structural model for p7 is that of an α-helical hairpin with two α-helical TMDs kinked in the middle (Cook et al. 2010), or a sequence divided into three helical segments (Ouyang et al. 2013) where the N-terminal half of the polypeptide would face the lumen of the channel and the C-terminal helix, p7(27–63), faces the lipid environment. The channel is formed by either six or seven monomers (Luik et al. 2009; Montserret et al. 2010).
p7-NS2 Interaction
Early genetic analyses suggested that p7 interacts with NS2, a polytopic membrane protein containing three N-terminal TMDs that is essential in the assembly process of the HCV particle (Jirasko et al. 2008). Mutation of residues in one protein to induce the emergence of complementary mutations in the other was used to identify the interaction network of NS2 protein with p7, E1 and E2, and NS3 proteins (Jirasko et al. 2010). Similar conclusions were reached in studies involving chimeric constructs with different genotypes (Pietschmann et al. 2006) which showed that virus release was most efficient when the N-terminal TMD of NS2 was from the same isolate as the core-to-p7 region. In a similar study, adaptive mutations in E1, p7, NS2, and NS3 were detected that were essential for virus assembly and/or release, again suggesting genetic interactions between these proteins (Yi et al. 2007).
Physical interaction between p7 and NS2 was observed during pull-down assays and mutagenesis (Ma et al. 2011), which suggested that p7 may regulate NS2-mediated complexes that are crucial for production of infectious HCV particles. Coimmunoprecipitation and FRET assays also supported a physical interaction between p7 and NS2 (Popescu et al. 2011), suggesting a complex between p7, NS2, and E2 mediated by transmembrane interactions. These interactions were proposed to be required to localize NS proteins and the core-containing cLDs to sites of virus assembly. Overall, these studies demonstrated that NS2, together with p7 protein, plays a central organizing role in HCV particle assembly by bringing together viral structural and nonstructural proteins. Although the exact mechanism linking nucleocapsid assembly with envelope acquisition is unknown, p7 and NS2 have been proposed to play a critical role in the migration of core protein and E1-E2 heterodimers to the virion assembly site (Vieyres et al. 2014).
Another role, in immune evasion, has been identified recently for p7 (Qi et al. 2017). Indeed, HCV acts against the host immune system by downregulating interferon (IFN) production, blocking IFN signaling transduction, and impairing IFN-stimulated gene (ISG) expression. But even when ISGs are expressed, most ISGs have been reported to be ineffective when overexpressed in virus-infected cells due to unknown mechanisms (Schoggins et al. 2011). By constructing a library of mutant HCVs with a 15-nt insertion, p7 was identified as an immune evasion protein that suppresses the antiviral IFN function, forming a complex with the host interferon-inducible protein 6–16 (IFI6–16) that has been verified by coimmunoprecipitation. It was proposed that while IFI6–16 acts to stabilize the mitochondrial membrane potential, p7 counteracts by depolarizing the mitochondrial potential, likely through its ion channel activity. Overall, the findings suggest that p7 antagonizes the antiviral responses of IFN by inhibiting the antiviral function of IFI6–16.
HCV Protein Interactome
A proteome-wide virus-host PPI screen of HCV was performed almost 10 years ago using a construction of a viral ORFeome and Y2H technology (De Chassey et al. 2008). In that study, 314 PPIs were identified involving viral and host proteins, 13 of which involving p7 and proteins expressed in the liver (Fig. 15.4), although these were not confirmed by orthogonal methods. A latter study combined mass spectrometry and functional genomics (Germain et al. 2014), but p7 was not included in the screen.

Graphical representation of the HCV virus-host (V-H) human interaction network (red lines) with black and red nodes representing viral and human proteins, respectively. Blue lines represent H-H interactions (adapted from De Chassey et al. (2008))
The interaction network between the ten HCV proteins has been investigated using a flow-cytometry-based FRET assay in living cells. In this study, p7 was found to bind NS2, Core, E1, and E2 (Hagen et al. 2014) (Fig. 15.5a). In 2016, a computational coevolution analysis of HCV attempted to reconstruct the PPI network of the HCV at the residue resolution (Champeimont et al. 2016). Coevolving residues were identified to predict PPIs for further experimental identification of HCV protein complexes (Fig. 15.5b). One of these interactions was p7-NS2 (see Fig. 15.5c).

(a) Network of reported intraviral HCV PPIs determined experimentally [adapted from Hagen et al. (2014)] compared to (b) coevolution links of HCV proteins (Champeimont et al. 2016), where blue lines correspond to coevolving links not experimentally reported; (c) predicted p7-NS2 interaction between F14 of NS2 and I19 in TM1 of p7 (red line), compared to experimentally reported interactions based on NMR (Cook et al. 2013) between NS2 A12 and V15 and W48 in p7 TM2 (green lines) [adapted from Champeimont et al. (2016)]
Coronavirus Viroporins
Coronaviruses (CoV) are vertebrate pathogens which cause human respiratory diseases that typically affect the respiratory tract and gut. CoVs belong to the family Coronaviridae, subfamily Coronavirinae, and are distributed into four genera α, β, γ, and δ (Enjuanes et al. 2000). While α-CoVs and β-CoVs circulate in mammalian hosts, γ-CoVs and δ-CoVs mainly infect birds. For example, β-CoVs include the murine hepatitis virus (MHV), whereas γ-CoVs include the avian infectious bronchitis virus (IBV). The first coronavirus was isolated in 1937 (IBV), whereas the first human coronavirus (HCoV) was identified in the 1960s. In humans, disease caused by coronaviruses ranges from mild to really severe, e.g., the recent severe acute respiratory syndrome (SARS) and the Middle East respiratory syndrome (MERS).
SARS-CoV appeared in 2002 causing ~10,000 human infections, with a 10% mortality rate (Holmes 2003). MERS coronavirus (MERS-CoV) emerged about 10 years later, and to date (as of July 2017) almost 2040 cases of infection and 712 deaths have been confirmed (http://www.who.int/emergencies/mers-cov/en/), i.e., a mortality of ~35%. Currently, no effective licensed prevention nor treatment exists against coronavirus infection (Lou et al. 2014), although live attenuated vaccines and fusion inhibitors are promising strategies.
CoVs have nonsegmented, exceptionally long genomes (up to 32 kb). One third of the genome hosts the ORFs for structural proteins, i.e., spike (S), envelope (E), membrane (M), and nucleoprotein (N). This part of the genome also encodes other so-called “accessory” proteins, which vary in number and sequence even among CoVs belonging to the same lineage (Enjuanes et al. 2008), e.g., from one in HCoV-NL63 to eight in SARS-CoV. The remaining two thirds of the genome encode nonstructural genes, with open reading frames ORF1a and ORF1b that produce polyproteins pp1a and pp1ab. These are then processed into 16 nonstructural proteins (nsp1 to 16); see Su et al. (2016) for recent general overview of coronaviruses and Forni et al. (2017) for the molecular evolution of HCoVs. In the case of SARS-CoV, the genome is predicted to encode 14 functional open reading frames, leading to the expression of up to 30 structural and nonstructural protein products.
E Viroporin
The E proteins are 76–109 amino acids long with one TMD (Torres et al. 2006; Li et al. 2014a; To et al. 2017), a short lumenal N-terminus and a longer cytoplasmic C-terminal tail (Nieto-Torres et al. 2011) which in SARS E protein tends to form β-structure in isolation, but it is mainly helical in the context of a full length protein (Li et al. 2014a). E proteins are localized particularly in the endoplasmic reticulum-Golgi intermediate compartment (ERGIC), where virus morphogenesis and budding occurs. E protein forms homopentameric channels with poor ion selectivity (Verdia-Baguena et al. 2012). Only the TMD of SARS-CoV E (E-TM) has been characterized in some detail, in lipid membranes (Torres et al. 2006) and in DPC micelles (Pervushin et al. 2009).
SARS-CoV E protein is a virulence factor critical for viral pathogenesis, as SARS-CoV lacking the E gene (rSARS-CoV-∆E) is attenuated in vivo (DeDiego et al. 2007). Mutations N15A and V25F abolish channel activity in vitro (Torres et al. 2007) and led to attenuation when introduced in a recombinant SARS-CoV (Nieto-Torres et al. 2014). The latter authors showed that channel activity is important for inflammasome activation and elevated production of the pro-inflammatory cytokine IL-1β. SARS-CoV E also regulates host stress response and apoptosis (DeDiego et al. 2011) and improves viral fitness. The importance of the E protein in coronavirus pathogenesis has led to the development of live attenuated vaccines based on E-deleted, E-truncated, or E-mutated virions, e.g., Regla-Nava et al. (2015). In general, E protein plays important roles in coronavirus assembly and morphogenesis, although E protein is not necessary to obtain infectious SARS-CoV (DeDiego et al. 2008).
E-M Interaction
E protein is a known binder of M protein, the most abundant protein component of the virion and the membrane protein responsible for its shape. M protein has three predicted TMDs and a large C-terminal extramembrane domain exposed to the cytoplasm or the interior of the virion. It is this domain that forms contacts with the C-terminal tail of the E protein, although TMD interactions are also likely (Lim and Liu 2001; Hogue and Machamer 2008). These interactions occur at the ERGIC, the budding compartment of the host cell. Since M-M interactions are major drivers of viral envelope formation, these contacts are likely to be important for particle assembly. The E-M interaction has long been reported in infectious bronchitis virus (IBV) and mouse hepatitis virus (MHV) by coimmunoprecipitation in virus-infected or virus-transfected cells (Corse and Machamer 2003; Lim and Liu 2001; Maeda et al. 1999) and was proposed to be crucial for the formation of virus-like particles (VLPs) and virions. In SARS-CoV, coexpression of M and E in a baculovirus expression system was sufficient for the assembly of VLPs (Ho et al. 2004). The deletion of the E gene (∆E) in the murine coronavirus, the mouse hepatitis virus (MHV), produced revertants where the M gene appeared to have been duplicated, creating new variants of M protein that lacked most of its C-terminal cytoplasmic tail (Kuo and Masters 2010). These results suggested a role for E proteins in “dispersing or de-aggregating” M protein during packaging.
Other CoV Viroporins
Although the most studied viroporin in CoVs is the envelope (E) protein, other viroporins have been found in an accessory gene present in all CoVs, between the S and E gene loci. In SARS-CoV (SARS-ORF3a) and in HCoV-229E (229E–ORF4a), these proteins form ion channels (Zhang et al. 2014; Lu et al. 2006), whereas HCoV-NL63-ORF3 has also been proposed to be a viroporin (Zhang et al. 2015). In HCoV-OC43, this genomic segment encodes ORF5, which has been reported to facilitate virion morphogenesis (Zhang et al. 2015). The latter has only a single TMD, in contrast with SARS-CoV ORF3a, HCoV-229E ORF4a, and HCoV-NL63 ORF3.
3a Viroporin
SARS-CoV 3a protein is a 274-amino-acid-long protein with three TMDs and forms homotetrameric complexes that have ion channel activity (Lu et al. 2006). 3a protein has a cysteine-rich domain (residues 127–133) responsible for homo- and hetero-dimerization (Lu et al. 2006). In addition to a Yxx domain and a diacidic domain, it has a C-terminal domain (Tan et al. 2006). The C-terminal domain has RNA-binding activity (Sharma et al. 2007). Protein 3a is suggested to play a structural role in the SARS-CoV life cycle, since it interacts with S, E, and M proteins (Tan et al. 2004) and it is incorporated into newly packaged matured SARS-CoV virions (Shen et al. 2005; Ito et al. 2005). Also, it regulates various cellular responses of host cells, e.g., the upregulation of fibrinogen gene expression (Tan et al. 2005), and the increase of IL-8 and NF-B promoter activities (Kanzawa et al. 2006), possibly through its RNA-binding activity (Sharma et al. 2007). SARS-CoV 3a protein induces caspase-dependent apoptosis both in vivo and in vitro (Wong et al. 2005).
Intraviral Interactions
Two yeast-two-hybrid (Y2H) studies have been conducted to study intraviral SARS-CoV protein interactions (von Brunn et al. 2007; Pan et al. 2008), although only a few of these interactions have been verified. Von Brunn et al. (von Brunn et al. 2007) reported interactions of E protein with the nonstructural proteins nsp1, nsp8, and nsp11, as well as with the accessory proteins ORF3b, ORF7b, and ORF9b, whereas ORF3a interacted with M and S. However, not all these interactions were confirmed by coimmunoprecipitation in mammalian cells. Overall, however, only 13% of the intraviral SARS interactions known at that time were identified. This is likely due to the bias of Y2H against membrane proteins, which prevents the transfer of expressed prey and/or bait fusion proteins to the nucleus to activate transcription. Pan et al. (Pan et al. 2008) also reported a genome-wide analysis of intraviral PPIs in SARS-CoV replication, using a mammalian two-hybrid system screen, although only two interactions of E and 3a were found here. In comparison with a similar screen in yeast, native posttranslational modifications and folding should be present, but the two methods share a similar limitation in terms of bias against membrane proteins.
Later, a tandem affinity purification (TAP) study coupled to mass spectrometry (Álvarez et al. 2010), using dual-tagged SARS-CoV E protein in infected cells as bait, identified viral proteins nsp3, S, and M and host proteins dynein heavy chain, fatty acid synthase, aminopeptidase puromycin sensitive, phosphofructokinase platelet, tubulin alpha and beta, actin beta, transmembrane protein 43, and lactate dehydrogenase A as binders. Nsp3 is the largest nonstructural protein of SARS-CoV (1922 amino acids) which is proposed to act as a replication/transcription scaffolding protein (Imbert et al. 2008). Interaction with nsp3 was confirmed by coimmunoprecipitation and was localized to one of the nsp3 seven domains, i.e., the N-terminal acidic domain (nsp3a), that has a ubiquitin-like fold (Serrano et al. 2007). Colocalization of E and nsp3 in the cytoplasm of the infected cell suggested nsp3 may bring E protein into the vicinity of the replication/transcription complex.
The PLpro domain of nsp3 has deubiquitinating activity (Lindner et al. 2005) and might act to protect the viral replication complex from proteasomal degradation via deubiquitination. The authors proposed that E-nsp3 interaction could control ubiquitination of E protein during infection. Interaction between nsp3a and SARS-CoV E was shown to involve residues in the C-terminal domain of the latter (Li et al. 2014a).
Host Interactions
More recently, two Y2H studies searched for host interacting partners using the C-terminal tail of SARS-CoV E as a bait (Teoh et al. 2010; Jimenez-Guardeño et al. 2014). The first of these reported the protein associated with Caenorhabditis elegans lin-7 protein 1 (PALS1) as a binder (Teoh et al. 2010). PALS1 is a tight junction-associated protein and part of a complex that maintains epithelial cell polarity (Fig. 15.6). Alterations of lung epithelia integrity were consistent with E protein hijacking PALS1 to the ERGIC/Golgi region. The E-PALS1 interaction is mediated by (i) a Postsynaptic density protein-95/Discs Large/Zonula occludens-1 (PDZ) domain present in PALS1 and (ii) the last four C-terminal residues of E protein which represent a putative type II PDZ-binding motif (PBM) (Harris and Lim 2001). PDZ domains are ~80–90 amino acids long and typically bind the C-terminal tails of proteins, although internal binding sites have also been reported [reviewed in Ye and Zhang (2013)]. They are usually found in signaling proteins that alter signaling pathways, with over 250 nonredundant PDZ domains being recognized in the human proteome (Wang et al. 2010).

Model of the potential consequences of SARS-CoV infection on polarity and intercellular junctions formed by alveolar epithelial cells. (a) The inner surface of human lung alveolae is lined with a monolayer of polarized epithelial cells. Components of CRB and PAR polarity complexes, including PALS1, are shown close to the apical domain. During infection, structural proteins accumulate in the ERGIC compartment, where SARS-CoV E could bind to PALS1 to disrupt its trafficking to the tight junction; (b) disruption of the tight junction and virus dissemination [adapted from Teoh et al. (2010)]
The C-terminal tail of SARS-CoV E protein, which includes the proposed PBM, forms a random coil secondary structure (Li et al. 2014a) in a variety of environments. However, PBMs usually fold as β-strands (Ye and Zhang 2013), which suggests that a β-structure conformation may be induced by target binding. Another similar Y2H study that used the same bait described a similar PDZ domain-containing binder, the syndecan-binding protein (syntenin) (Jimenez-Guardeño et al. 2014). Syntenin is a scaffolding protein that can initiate a signaling cascade resulting in the phosphorylation and activation of p38 mitogen-activated protein kinase (p38-MAPK), leading to expression of pro-inflammatory cytokines. The authors showed that the proposed C-terminal PBM in SARS-CoV E protein is a determinant of virulence. Since SARS-CoV-infected patients show an exacerbated inflammatory response that leads to epithelial and endothelial damage, edema, and acute respiratory distress syndrome (ARDS), the disruption of this pathway may have therapeutic implications. Overall, an involvement of this PBM in E protein in epithelial integrity and inflammatory responses is likely. Several other viruses, e.g., influenza A virus or human papillomavirus, have been described to enhance pathogenesis through viral proteins containing PBMs [reviewed in Javier and Rice (2011)], which probably constitute a common viral strategy.
CoV E proteins may also interact with, and modulate, host channels to support the virus life cycle. In Xenopus oocytes, it has been shown that coexpression of SARS-CoV E with human epithelial sodium transporter (ENaC) reduced amiloride-sensitive current through PKC activation followed by reduction of ENaC surface levels (Ji et al. 2009). A similar direct or indirect inhibitory effect on other endogenous channels was proposed from patch-clamp experiments using SARS-CoV E-transfected cells (Nieto-Torres et al. 2011). For IBV E, interaction with endogenous channels or SNAREs has been suggested to justify the Golgi complex rearrangement in response to IBV E expression (Ruch and Machamer 2011), although this observation may also involve the IBV E channel itself. For example, ion homeostasis at the Golgi could affect Na+/H+ exchangers that are critical for maintaining low luminal pH. Interactions of viroporins with Golgi channels or transporters are largely unexplored in the viroporins field, but notable cases have been already reported. For example, oncogenic protein E5 from papillomavirus (Wetherill et al. 2012) is able to bind the 16 K subunit of the lumen-acidifying V-ATPase (Goldstein et al. 1991), preventing assembly of the pump and leading to alkalinization of the Golgi lumen (Schapiro et al. 2000).
The Respiratory Syncytial Virus Small Hydrophobic Protein (RSV-SH)
Human respiratory syncytial virus (hRSV) belongs to the Paramyxoviridae family in the pneumovirus genus. This enveloped virus has a negative-sense single-strand RNA genome 15.2 kb long that encodes 10 sub-genomic mRNAs and 11 proteins (Fields et al. 2013). These 11 proteins include three membrane proteins accessible to the surface of the virion: the two that generate most RSV-neutralizing antibodies, fusion (F) and attachment (G), and the small hydrophobic (SH) protein.
RSV affects more than 30 million children below 5 years old and is the leading cause of bronchiolitis and pneumonia in infants and elderly (Dowell et al. 1996). Disease caused by RSV is responsible for 200,000 deaths worldwide which mostly occur in developing countries. hRSV exists as two antigenically distinct subgroups, A and B, both capable of inducing severe lower respiratory tract (LRT) disease in humans (Hall et al. 1990).
Although the virus was isolated more than half a century ago, no effective licensed treatment or vaccine is available for the general population, despite promising RSV vaccine candidates in clinical trials. Palivizumab is a humanized monoclonal antibody (IgG) directed against the F protein that is recommended for infants <2 years old with high risk. However, it is not effective therapeutically and is only moderately effective at preventing infection. Since it costs $4500 per treatment course (Weiner et al. 2011), its use is limited to a small fraction of patients worldwide. The only licensed drug for therapeutic use is a nucleoside analog which has limited efficacy.
SH Viroporin
The SH protein in hRSV is only 64 (subgroup A) or 65 (subgroup B) amino acids long, but its sequence is well conserved, especially the N-terminal extramembrane domain (Tapia et al. 2014). It has a single TM α-helical hydrophobic region, with C- (lumenal or extracellular) and N- terminal (cytoplasmic) extramembrane domains (Collins and Mottet 1993). The N-terminal cytoplasmic domain forms a short α-helix (residues 5–14) (Fig. 15.7a), almost coincident with a “10-residue” conserved sequence between hRSV and MuV SH protein sequences. SH proteins in MuV, PIV5, and JPV have extremely short lumenal domains (nine, two, and ten residues, respectively) compared with their much longer N-terminal cytoplasmic domains, which are likely involved in PPIs. The C-terminal extramembrane domain forms an extended β-hairpin. In bicelles, the α-helix of the TMD extends up to residue His-51 (Li et al. 2014b), resulting in both protonatable residues of SH protein, His-22, and His-51, oriented toward the lumen of the channel.

Structural model of SH protein monomer. (a) Comparison of models of monomeric SH protein obtained in micelles (red) and in bicelles (blue), with residues prolonging the TM domain up to His-51 (Li et al. 2014b); (b) residues in SH involved in interaction with BAP31; N-terminal cytoplasmic helix of SH protein, with residues perturbed (red) after addition of BAP31 cytoplasmic domain to labeled SH protein in detergent micelles (Li et al. 2015)
SH protein forms homo-oligomers (pentamers), and this oligomeric form is responsible for ion channel (IC) activity (Gan et al. 2012; Gan et al. 2008) that has poor ion selectivity. In infected cells, most SH protein accumulates at the membranes of the Golgi complex, but it is also found in the ER or plasma membrane (Rixon et al. 2004). SH has potential glycosylation sites in both the C- and N-terminal domains (Collins et al. 1990). In infected cells, the SH protein of strain A2 accumulates in four different forms (Olmsted and Collins 1989; Collins et al. 1984; Collins and Mottet 1993), but the most abundant is a full-length unglycosylated form. The G protein forms G-F and G-SH complexes, but direct interactions between SH and F have not been observed (Low et al. 2008).
SH and apoptosis. It has been proposed that SH protein blocks apoptosis through inhibition of the TNF-α pathway (Fuentes et al. 2007), but the mechanism of this inhibition is not clear. A similar anti-apoptotic effect of SH protein has been reported for other members of the Paramyxoviridae family that encode SH proteins, e.g., mumps virus (MuV) and the parainfluenza virus 5 (PIV5).
Incidentally, an anti-apoptotic effect has also been noted for other similar viral channels (viroporins), e.g., E5 in the human papillomavirus type 16 (HPV-16) (Kabsch et al. 2004), or the envelope (E) protein, a viroporin in the severe acute respiratory syndrome (SARS) virus (DeDiego et al. 2011).
SH and the Inflammasome
SH protein is also involved in inflammasome regulation, but the mechanism involved is not known. Indeed, some authors have proposed that RSV SH has a role in regulation of the NLRP3 inflammasome (Russell et al. 2015). The latter is “primed” after the recognition of viral genomic RNA (vRNA) by pattern recognition receptors (PRRs) and subsequent activation of NF-kB. This priming involves the expression of inflammasome components, e.g., NLRP3 and inactive procaspase-1 (Elliott and Sutterwala 2015). Various virus-induced damage-associated molecular patterns (DAMPs) induce the assembly and activation of the NLRP3 inflammasome. This leads to processing of procaspase-1 into active caspase-1, which in turn cleaves inactive pro-IL-1β into the mature form IL-1β. The latter is a potent pro-inflammatory cytokine crucial in resolving infectious processes.
Various viruses can activate the inflammasome by disrupting ion homeostasis through the expression of viroporins. For example, influenza A virus (IAV) activates NLRP3 as a result of H+ or ion flux from Golgi mediated by the M2 channel (Ichinohe et al. 2010). The 2B protein in picornaviruses induce NLRP3 cytoplasmic relocalization and inflammasome activation in an intracellular Ca2+-mediated manner (Ito et al. 2012), while a similar mechanism has been proposed for SARS-CoV E (Nieto-Torres et al. 2015). The latter triggered inflammation in the lungs of mice, leading to epithelial cell damage and death (Nieto-Torres et al. 2014), and this was correlated to high levels of mature IL-1β in the airways of infected animals.
Similarly, RSV SH protein has been suggested to activate the NLRP3 inflammasome through its IC activity and ion leakage from the Golgi (Triantafilou et al. 2013), similar to the mechanism proposed for SARS-CoV E (Nieto-Torres et al. 2015). Another study (Russell et al. 2015) proposed that IL-1β is overproduced when SH is absent from RSV. The study also showed attenuation in mice when infected with RSV ΔSH. That deletion of RSV SH leads to an increase in IL-1β is also supported by studies in bovine RSV (bRSV), where a ΔSH vaccine strain induced higher levels of IL-1β in the lungs of infected cattle (Taylor et al. 2014). Consistent with this, lung macrophages infected with RSV did not lead to increased IL-1β, although other pro-inflammatory cytokines were overexpressed (Ravi et al. 2013). Overall, it has been proposed (Russell et al. 2015) that SH protein blocks IL-1β production, preventing the clearance of infected cells. Indeed, blockade of IL-1β prior to infection increased the viral load, supporting the idea that SH might enable immunomodulation. The interaction between SH and G protein has also been shown previously to have an immunomodulatory role (Polack et al. 2005).
Although in cell culture RSV ΔSH is still viable, grows to similar titer to wild-type RSV, and still forms syncytia, SH-deleted RSV (RSV ΔSH) is significantly attenuated in a variety of hosts (Taylor et al. 2014; Bukreyev et al. 1997; Russell et al. 2015). Indeed, in the last few years, one of the leading RSV LAVs have included, among other modifications, a deletion in the SH gene (Karron et al. 2005). The cause of attenuation is not known, although it may have to do with effective transmission of the virus (Bukreyev et al. 1997).
A transcriptome analysis comparing RSV with and without SH protein could help to decipher the role of SH during infection and the cause of attenuation in vivo and to associate these responses to specific SH domains or features.
Host Interactions
Recently, a membrane-based yeast two-hybrid system (MbY2H) was used to identify a cellular binding partner of hRSV SH protein, the B-cell receptor-associated protein 31 (BAP31) (Li et al. 2015), in a human lung cDNA library. BAP31 is a membrane protein located at the ER that has an essential role in sorting newly synthesized membrane proteins. Additionally, BAP31 has a cytoplasmic C-terminus with two coiled coils (Quistgaard et al. 2013), one of them containing a variant of the death effector domain (vDED) flanked by two caspase-8 cleavage sites. This domain is excised upon activation of caspase-8 to produce a fragment p20, known to function as a proapoptotic factor (Breckenridge et al. 2003). This interaction was confirmed using co-transfection, pull-down assay and immunofluorescence colocalization, and also using endogenous BAP31 and was localized to the first N-terminal 44 residues (Li et al. 2015). When 15N–labeled SH protein was titrated with cytoplasmic C-terminal domain of human BAP31, major shifts were observed at residues I6, I8, S12, and W15 (Fig. 15.7b). It can be hypothesized that this interaction could interfere with the interaction between BAP31 and caspase-8, blocking the cleavage sites and preventing conversion to the pro-apoptotic form of BAP31, i.e., p20, thus delaying apoptosis. Incidentally, the viroporin E5 from the high-risk human papillomavirus HPV-16 and HPV-31 was also found to interact with BAP31, where it is similarly thought to regulate apoptosis in addition to its roles in immunomodulation (Regan and Laimins 2008) (see below).
Intraviral Interactions
The interaction between RSV SH and G proteins has been reported previously in infected cells (Low et al. 2008; Rixon et al. 2005), although its significance is not yet clear. F protein seems to be the main determinant of host cell specificity during virus entry, and both F and G proteins are able to bind heparin sulfate, the putative cell receptor for RSV (Kargel et al. 2001). However, a tri-component complex between SH, G, and F proteins was not observed (Low et al. 2008). Both G and F proteins have one predicted TMD, and interaction with SH protein can be both through the TMDs or extramembrane domains of the latter.
Until now, all studies to determine the role of SH protein in RSV infection have used wild-type RSV and ∆SH RSV (SH gene deleted) and compared the effects of this deletion on various parameters in infected cells, or in animal models. The effects caused by transfection of SH protein in readouts that depend on, for example, inflammasome activation or apoptosis, have also been explored. However, comprehensive datasets that aim at elucidating the contribution of SH to virulence observed in vivo, and a rationale for the attenuation observed when SH is deleted, are lacking.
The Human Immunodeficiency Virus Viral Protein U (HIV-1-Vpu)
The human immunodeficiency virus type 1 (HIV-1) is an enveloped virus that causes AIDS. It has a single-stranded, positive-sense RNA genome of 9.8 kb which encodes for nine genes: the structural genes gag, pol, and env, the regulatory genes tat and rev, and additional genes nef, vif, vpr, and vpu which encode for accessory proteins. One of these accessory proteins, the Vpu (viral protein U) (Cohen et al. 1988), is an 81-residue small-membrane protein consisting of an N-terminal transmembrane helix and a C-terminal cytoplasmic domain which contains two helices linked by a flexible loop region [reviewed in Opella (2015)] (Fig. 15.8a). Vpu can oligomerize to form cation-selective channels in membranes, although the rationale for this channel activity is not well defined.

(a) Predicted secondary structure of Vpu showing N-terminal TMD (blue) and two α-helices of the cytoplasmic (CYTO) domain (red). Phosphorylated S52 and S56 are represented as circles (adapted from Dubé et al. (2010)). (b) Solution NMR structure of VpuCYTO in DPC micelles (Protein Data Bank code: 2K7Y). Helix 1 (Ile39-Glu48) and helix 2 (Leu64-Arg70) are shown as ribbons. β-TrCP-binding DSGxxS motif is in blue. Side chains of phosphorylated serines are shown as balls and sticks. VpuCYTO residues showing substantial chemical shift changes upon addition of 1 mM CD4mut are in red (adapted from Singh et al. (2012)); (c) Vpu-mediated degradation of CD4. Binding of Vpu to CD4 is mediated by the cytoplasmic helices and TM helices. Vpu has two conserved phosphoserines which constitute a binding motif for β-TrCP, leading to assembly of an E3 ubiquitin ligase (UbL) complex that results in CD4 ubiquitination and proteasomal degradation (modified from Strebel (2014))
Vpu has two primary roles during HIV-1 infection: (i) enhancement of virion release (Terwilliger et al. 1989; Strebel et al. 1988) and (ii) degradation of host CD4 receptor (Willey et al. 1992). Absence of Vpu in infected cells correlates with reduction of viral release and intracellular accumulation of HIV-1 viral proteins (Klimkait et al. 1990). The host protein BST-2 is a restriction factor of HIV-1 release, and its activity can be neutralized by Vpu (Neil et al. 2008; Van Damme et al. 2008).
Host Interactions
A number of host factors have been reported to bind Vpu, including the immunoreceptors major histocompatibility complex class I (MHC-I) (Kerkau et al. 1997), CD1d (Moll et al. 2010), NK-T-B-antigen (NTB-A) (Shah et al. 2010), poliovirus receptor (PVR) (Matusali et al. 2012) and human leukocyte antigen C (HLA-C) (Apps et al. 2016), C-C chemokine receptor type 7 (CCR7) (Ramirez et al. 2014) and CD62L (Vassena et al. 2015), tetraspanins (Haller et al. 2014; Lambelé et al. 2015), K+ channel TASK-1 (Hsu et al. 2004), metabolic transporter, sodium-coupled neutral amino acid transporter 1 (SNAT1) (Matheson et al. 2015), and the most recently reported intercellular adhesion molecule 1 (ICAM-I) (Sugden et al. 2017). However, the most important binders are CD4 and BST-2.
Vpu-Mediated Degradation of CD4
The host CD4 is a cell surface receptor critical for HIV-1 entry into target cells by endocytosis, but its expression at the cell surface prevents the release of infectious virions from infected cells. To counteract this host defense mechanism, viral Vpu, Env, and Nef downmodulate CD4 surface expression [see review in Doms and Trono (2000)]. Vpu interacts with newly synthesized CD4 at the ER to mediate CD4 degradation (Willey et al. 1992). Vpu-mediated degradation of CD4 requires physical interaction between the two proteins. In CD4, the interaction domain has been mapped to a specific motif (L414SEKKT419) and a membrane-proximal α-helix (Bour et al. 1995). For Vpu, residues in both cytoplasmic α-helices of Vpu experienced chemical shift perturbations upon CD4 binding (Fig. 15.8b) (Singh et al. 2012), although involvement of the TMDs of both proteins has also been suggested (Magadán and Bonifacino 2012; Do et al. 2013). However, the scrambling or replacement of the whole Vpu TMD appears to have no effect on Vpu-mediated degradation of CD4 (Willey et al. 1994; Schubert et al. 1996), whereas a single amino acid substitution at the TMD, W22 L, abolished CD4 degradation but did not disrupt the CD4-Vpu interaction (Magadán and Bonifacino 2012), suggesting that TM interactions between CD4 and Vpu may function beyond the expected role of stabilizing the protein complex for CD4 degradation.
Overall, it has been proposed that Vpu acts as an adapter protein to link CD4 to the host ubiquitin-proteasome machinery for degradation. This binding triggers the recruitment of the host beta-transducin repeat-containing protein (β-TrCP). A conserved di-phosphoserine motif (D51SGxxS56) located within the loop region that connects the two cytoplasmic α-helices of Vpu is necessary for this process. Interaction of the phosphoserines in Vpu with the WD boxes of β-TrCP enables the formation of a CD4-Vpu-β-TrCP ternary complex (Margottin et al. 1998), bringing CD4 and other components of the SCFβ-TrCP E3 ubiquitin ligase complex (Skp1 and Cullin1) in close proximity to facilitate the trans-ubiquitination of the CD4 cytosolic tail (Binette et al. 2007) on lysine and serine/threonine residues (Magadán et al. 2010) and subsequently its transportation to the cytosol for degradation (Fig. 15.8c).
Vpu-Mediated Antagonism of BST-2
Vpu enhances virus dissemination by antagonizing the host BST-2, a host restriction factor with antiviral capabilities [reviewed in Simon et al. (2015)]. BST-2 is an interferon-inducible type II integral membrane protein located at the budding site of HIV-1. BST-2 has an N-terminal cytoplasmic domain, a TMD, followed by a coiled-coil ectodomain and finally a C-terminal glycosyl-phosphatidylinositol (GPI) membrane anchor. Its unusual topology enables it to tether virions by inserting, preferentially its GPI anchor, into the envelope of assembling virion particles, while itself remains embedded in its host cell membrane (Venkatesh and Bieniasz 2013; Neil et al. 2006).
It has been proposed that Vpu engages BST-2 through interaction between their respective TMDs, with involvement of Vpu’s A14, W22, and A18 (Vigan and Neil 2010) and BST-2’s I34, L37, and L41 (Kobayashi et al. 2011). An NMR study described an antiparallel interaction between Vpu and BST-2 TMDs in DHPC micelles, in an orientation where A18 of Vpu faces L37 of BST-2 (Skasko et al. 2012) (Fig. 15.9). The conserved residues within the Vpu membrane-proximal cytoplasmic hinge region (E28YRKIL33) have also been found to be important for Vpu-mediated BST-2 antagonism (Lukhele and Cohen 2017).

Left, the spin label (MTSL) at the N-terminus of Vpu selectively decreased the intensity of C-terminal residues of BST-2, whereas the spin label at the N-terminus of BST-2 selectively decreased the intensity of C-terminal residues of Vpu; right, helical wheel diagrams of the BST-2 and Vpu TMDs. The TMDs of BST-2 and Vpu are depicted in their anti parallel orientation (adapted from Skasko et al. (2012))
The mechanism of BST-2 neutralization by Vpu appears to be multifaceted and under debate, although the key mechanism appears to be the direct displacement of BST-2 from the virus assembly sites at the plasma membrane (McNatt et al. 2013). Accordingly, a C-terminal Trp residue at the Vpu cytoplasmic tail has been reported to contribute to this displacement of cell surface BST-2 (Lewinski et al. 2015). Other proposed mechanisms have been proposed, e.g., Vpu can disrupt intracellular BST-2 trafficking by sequestering both newly synthesized and recycling BST-2 within intracellular compartments such as the TGN (Dubé et al. 2010; Hauser et al. 2010). This effectively blocks the resupply of BST-2 to the plasma membrane and thereby reduce BST-2 surface density (Dubé et al. 2011). Vpu-mediated BST-2 mistrafficking has been reported to involve the host clathrin-dependent membrane trafficking pathways which are mediated by clathrin adaptor protein (AP) complexes (Kueck and Neil 2012; Lau et al. 2011). It was proposed that Vpu is able to hijack the clathrin-dependent trafficking machinery via a mimicked canonical acidic di-leucine sorting motif (E59xxxLV64) within its second cytoplasmic α-helix to recruit the AP complexes, forming a Vpu-BST-2-AP ternary complex (McNatt et al. 2013; Jia et al. 2014; Kueck et al. 2015). In addition, the conserved di-phosphoserine motif (D51SGxxS56) in Vpu may also be required for this recruitment (Kueck et al. 2015).
A crystal structure of a VpuCYTO-BST-2CYTO fusion protein in complex with the AP1 core has been solved (Jia et al. 2014). In this model, the cytoplasmic domains of Vpu and BST-2 do not interact directly. Instead, Vpu seems to act as a chaperone to enhance binding of AP1 to BST-2. Stability of this complex is achieved by pair-wise binary interactions between Vpu and BST-2 TMDs and between Vpu and BST-2 cytoplasmic domains to several parts of AP1 (Fig. 15.10). Thereafter, the Vpu-BST-2 complex is thought to proceed through the clathrin-mediated trafficking pathway for β-TrCP-dependent ubiquitination of BST-2 before subsequent ESCRT-mediated endo-lysosomal degradation.

Schematics of Vpu hijacking of AP1 to target BST2. AP1 is colored by subunit (β1 in gray, γ in orange, μ1 in green, and σ1 in yellow). VpuCYTO (cyan) binds to the acidic di-leucine-binding pocket of γ/σ1, and BST-2CYTO (magenta) binds to the tyrosine-binding pocket in μ1. Transmembrane helices are represented by cylinders (adapted from Jia et al. (2014))
Recently, it has been reported that Vpu may hijack the function of the host actin cross-linking regulator filamin A (FLNa) during its quest of BST-2 modulation (Dotson et al. 2016). Vpu may also exploit an LC3-associated noncanonical autophagy pathway to restrict BST-2 (Madjo et al. 2016). The refinement of current models, together with the discovery of additional host and/or intraviral factors involved, will ultimately form a complete and accurate picture of Vpu-mediated BST-2 antagonism.
The Polyomavirus JC Agnoprotein
Polyomaviruses are small, non-enveloped DNA viruses with their closed circular genome packaged within an icosahedral viral capsid. Progressive multifocal leukoencephalopathy (PML), a deadly demyelinating disease of the brain, is attributed to the human polyomavirus JCV (John Cunningham virus). JCV encodes for a small and highly basic protein called agnoprotein which has important regulatory roles in the JCV replication cycle. Besides JCV, the agnoprotein can also be found in other polyomaviruses including BK virus (BKV) and simian virus 40 (SV40) (Sariyer et al. 2011). The 71-residue agnoprotein has a central hydrophobic region which has been reported to form an amphipathic α-helix (Lys23-Phe39) (Coric et al. 2014). This helix is characteristically rich in Leu/Ile/Phe, which is required for protein stability and oligomerization (Saribas et al. 2013). A recently solved NMR structure in organic solvent has revealed a second α-helix, albeit minor, spanning residues Leu6-Lys13 (Fig. 15.11) (Coric et al. 2017).

NMR structure of agnoprotein contains two main α-helical structures (red) and two unstructured regions (yellow) [adapted from Coric et al. (2017)]
Agnoprotein demonstrates several key features which are commonly shared among viroporins (Suzuki et al. 2010a; Suzuki et al. 2013; Suzuki et al. 2010b), e.g., it is a membrane protein that associates into homo-oligomers that increased membrane permeability leading to influx of extracellular Ca2+ and enhancement of virus release. In addition, agnoprotein-deleted mutants have defective virion release and viral propagation.
Intraviral and Host Interactions
The JCV agnoprotein has been shown to interact with a number of viral proteins: large T-antigen (LT-ag) (Safak et al. 2001), small t-antigen (St-ag) (Sariyer et al. 2008), HIV-1 Tat (Kaniowska et al. 2006), and capsid protein VP1 (Suzuki et al. 2012). It also interacts with cellular proteins, including the Y-box-binding factor 1 (YB-1) (Safak et al. 2002), tumor suppressor p53 (Darbinyan et al. 2002), tubulin (Endo et al. 2003), DNA damage repair protein Ku70 (Darbinyan et al. 2004), fasciculation and elongation protein zeta 1 (FEZ1) (Suzuki et al. 2005), heterochromatin protein 1 alpha (HP-1α) (Okada et al. 2005), protein phosphatase 2A (PP2A) (Sariyer et al. 2008), and the adaptor protein complex 3 (AP3) δ subunit (Suzuki et al. 2013).
One of the interesting host factors targeted by the JCV agnoprotein is the AP3 (Suzuki et al. 2013). Interaction of agnoprotein with the δ subunit of AP3 (AP3D) appears to hijack the AP3-mediated intracellular vesicular trafficking to prevent the targeted lysosomal degradation of agnoprotein. This phenomenon is reminiscent of the special features of viroporins such as M2 and Vpu, which also manipulate host trafficking pathways. Agnoprotein is then allowed to be translocated to the plasma membrane to act as a viroporin and promote virion release (Suzuki et al. 2013). Alanine substitutions of two basic residues (Arg8 and Lys9) in the N-terminus of agnoprotein disrupt its viroporin activity (Suzuki et al. 2010a) and also disrupt binding to AP3D, ensuing its transport to the lysosomes and subsequent lysosomal degradation (Fig. 15.12) (Suzuki et al. 2013). These basic residues are part of an ordered helical structure (Coric et al. 2017) and may constitute an important regulatory and/or interaction motif.

Involvement of AP-3 in membrane permeabilization and virion release involving WT (left) and mutant RK8AA (right) agnoprotein. Both WT and RK8AA form homo-oligomers as integral membrane proteins in cytoplasmic organelles. WT disrupts AP3-mediated vesicular trafficking, is translocated to plasma membrane, and functions as a viroporin, resulting in the promotion of virion release. In contrast, the RK8AA mutant fails to bind to AP3D and does not disrupt AP3-mediated vesicular trafficking and is transported to lysosomes and degraded. RK8AA agnoprotein cannot promote virion release and is defective in viral propagation [adapted from Suzuki et al. (2013)]
Rotavirus NSP4
Rotaviruses are members of the Reoviridae family of non-enveloped viruses which consist of segmented, double-stranded RNA genomes surrounded by multiple concentric protein capsids (Coombs 2006). Rotaviruses are a leading cause of severe viral gastroenteritis and dehydrating diarrhea in infants and young children, resulting in a high global child mortality rate of 215,000 in 2013 (in children <5 years old) (Tate et al. 2016). The rotavirus nonstructural protein 4 (NSP4) is a 175-amino-acid transmembrane ER glycoprotein and the first virus-encoded enterotoxin to be discovered (Ball et al. 1996). Besides its primary ER localization, NSP4 can also be secreted as a soluble enterotoxin (Bugarc̀ić and Taylor 2006) or colocalize with the autophagy protein LC3 in cap-like structures that associate with viroplasms (Berkova et al. 2006).
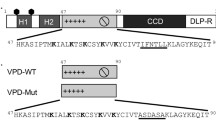
NSP4 consists of three hydrophobic domains (H1, H2, and H3) followed by a long cytoplasmic region containing a coiled-coil domain (CCD) and a flexible tail region (Estes and Greenberg 2013) (Fig. 15.13). Notably, its distinctive functional domains include (i) an enterotoxic domain (ETD, residues 114–135), which can function as a diarrhea-inducing enterotoxin similar to the full-length protein in young mice (Ball et al. 1996), and (ii) a recently discovered viroporin domain (VPD, residues 47–90) which is made of a penta-lysine domain and the amphipathic helix H3 (Hyser et al. 2010) and exhibits cation-selective channel activity in artificial lipid bilayers (Pham et al. 2017).

Schematic representation of rotavirus NSP4 structural domains. H1, H2, and H3, hydrophobic domains 1, 2, and 3, respectively. NSP4 viroporin domain (residues 47–90) and enterotoxic domain (residues 114–135) are indicated. Putative binding sites of NSP4 interaction partners are also indicated
An alteration in cellular calcium homeostasis is critical for rotavirus replication and cytopathogenesis, and this has been correlated with the NSP4 viroporin [see review in Hyser and Estes (2015)]. Earlier studies reported that NSP4 colocalize with the autophagosome marker LC3 in “cap-like structures” associated with viroplasms (Berkova et al. 2006), sparking interest of whether the host autophagy machinery is manipulated during rotavirus infection. Indeed, NSP4 appears to orchestrate a series of events which ultimately lead to host autophagy. NSP4 viroporin activity at the ER can activate the ER calcium sensor stromal interaction molecule 1 (STIM1), which triggers an activation of store-operated calcium entry (SOCE), which in turn facilitates Ca2+ influx through the plasma membrane (Hyser et al. 2013). This increase in intracellular Ca2+ activates the Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKK-β) to initiate autophagy (Anderson et al. 1999; Crawford et al. 2012). The autophagy membrane trafficking pathway is then hijacked by the virus to transport viral proteins from the ER to viroplasms for assembly of infectious virus (Crawford et al. 2012).
Intraviral and Host Interactions
Even before the discovery of its viroporin activity, NSP4 has been described to perform multiple functions through interacting with a number of viral and host factors, and the cytoplasmic region of NSP4 is an interaction hotspot. For instance, the NSP4 cytoplasmic CCD has been reported to be the binding site for the rotavirus spike protein VP4 (NSP4 aa112–148) (Au et al. 1993), host extracellular matrix (ECM) proteins laminin-β3 and fibronectin (NSP4 aa87–145) (Boshuizen et al. 2004), caveolin-1 (Cav-1) (NSP4 aa114–135) (Parr et al. 2006; Ball et al. 2013), and the integrin I domains (NSP4 aa114–130) (Seo et al. 2008). In addition, the flexible region of the NSP4 cytoplasmic tail interacts with VP6 to serve as an intracellular receptor for the viral double-layered particles (DLPs) (NSP4 aa161–175) (Au et al. 1989; Taylor et al. 1996) and can also bind microtubules (NSP4 aa120–175) (Xu et al. 2000). NSP4 can also bind the host calnexin via the two N-linked high-mannose oligosaccharide residues within the NSP4 H1 domain (Mirazimi et al. 1998). The putative binding sites for these interactions are summarized (Fig. 15.13).
While mechanistic and structural information on membrane insertion and oligomerization of the full-length NSP4 is still lacking, a topology model of NSP4 as a three-pass transmembrane protein has been proposed (Fig. 15.14). In addition, crystal structures have revealed that the NSP4 CCD from two different rotavirus strains can form a tetramer and a pentamer, respectively (Bowman et al. 2000; Chacko et al. 2011) and that the tetrameric NSP4 CCD, but not the pentameric form, can bind Ca2+ at its core. Later studies clarified that the oligomeric status of NSP4 CCD can be regulated by pH and Ca2+; at neutral pH it forms a tetramer that binds Ca2+, but at low pH it forms a pentamer that does not bind Ca2+ (Sastri et al. 2014). While the NSP4 CCD appears to be an interaction hotspot, it remains to be revealed how environmental cues may influence its binding conformations and affinity with interaction partners. For instance, the CCD may act as a cytoplasmic pH/Ca2+ sensor that alters the NSP4 oligomeric state and conformation to regulate its binding to a certain interaction partner, or Ca2+ may even act as a cofactor for binding.

Model of NSP4 as a three-pass transmembrane protein. Left: initial ER membrane insertion of NSP4 mediated by uncleaved signal sequence in H2 domain. Lysine residues interact with membrane phospholipid, promoting insertion of the viroporin domain (PD + AD) as an anti-parallel α-helical hairpin. Center: insertion of the viroporin domain generates a three-pass transmembrane topology. Right: NSP4 oligomerization around the amphipathic α-helix generates an aqueous pore for the passage of ER Ca2+ [adapted from Hyser et al. (2010)]
Human Papillomavirus E5
Human papillomaviruses (HPVs) are double-stranded DNA viruses which are small, non-enveloped, and epitheliotropic. They are known to infect mucosal and cutaneous epithelia of the anogenital tract and the hands/feet regions. High-risk HPVs, mainly HPV-16 and HPV-18, are responsible for 70% of cervical cancers and precancerous cervical lesions. The HPV-16 E5 (16E5), a small hydrophobic oncoprotein, is a recent addition to the viroporin family as it exhibits ion channel activity in vitro (Wetherill et al. 2012). 16E5 is an 83-residue protein with three putative TMDs and an N-terminal luminal, C-terminal cytoplasmic topology (Krawczyk et al. 2010). 16E5 monomers oligomerize as homodimers or hexamers (Kell et al. 1994; Gieswein et al. 2003; Wetherill et al. 2012). 16E5 has roles in cellular transformation, mitogenic signaling, immune evasion, intracellular protein trafficking, and apoptosis [reviewed in Müller et al. (2015)].
Host Interactions
It has been reported that 16E5 forms a stable complex with the epidermal growth factor receptor (EGFR) in co-transfected cells (Hwang et al. 1995) and with the 16 K subunit of the vacuolar H+-ATPase (V-ATPase) (Conrad et al. 1993), although the 16E5 binding site with the latter is under debate (Adam et al. 2000; Rodríguez et al. 2000). The C-terminal domain of 16E5 has been reported to bind the nuclear transport receptor karyopherin β3 (KNβ3) (Krawczyk et al. 2008) and the Ca2+/phospholipid-/actin-binding protein calpactin I (Krawczyk et al. 2011). Other reported interaction targets of 16E5 include the gap junction protein connexin 43 (Oelze et al. 1995; Tomakidi et al. 2000), growth factor receptor ErbB4 (Chen et al. 2007), zinc transporter ZnT-1 (Lazarczyk et al. 2008), transmembrane channel-like proteins EVER1 and EVER2 (Lazarczyk et al. 2008), the putative ER ion channel A4 (Kotnik Halavaty et al. 2014), and the Golgi-resident transmembrane protein YIPF4 (Müller et al. 2015).
Interactions between 16E5 and host proteins have been implicated in the modulation of host defense. For instance, 16E5 can help HPV escape from immunesurveillance by downregulating expression of antigen-presenters at the host cell surface. 16E5 binds and retains MHC-I in the ER and Golgi to prevent its trafficking to the cell surface, in an interaction involving two leucine pairs in the first TMD (TM1) of 16E5 and the heavy chain of MHC-I (Ashrafi et al. 2005; Cortese et al. 2010; Ashrafi et al. 2006). The TM1 of 16E5 may also bind and cripple the function of the MHC-I chaperone, Bap31 (Ladasky et al. 2006; Regan and Laimins 2008; Cortese et al. 2010). Intriguingly, a motif consisting of ten identical residues between the 16E5 TM1 and Bap31 TM3 has been discovered (Fig. 15.15) and could represent a case of molecular piracy used by 16E5 to displace Bap31 from MHC-I. In addition, 16E5-mediated ER retention of MHC-I may also involve an interaction with the ER chaperone calnexin, since surface downregulation of MHC-I is not observed in calnexin-deficient cells (Gruener et al. 2007). In the same study, a tri-protein complex of 16E5, MHC-I, and calnexin could be obtained based on a coimmunoprecipitation assay. The 16E5-calnexin interaction also reduced CD1d surface levels by retaining it in the ER and subsequently redirecting it for proteasomal degradation (Miura et al. 2010).

E5 and BAP31 topology. Top, topology of E5 and Bap31, with TMDs indicated; TM3 in BAP31 and TM1 in 16E5 share a similar motif [adapted from Cortese et al. (2010)]. The alignment of 16E5 TM1 and Bap31 TM3 showing the ten-residue identity which may be involved in interactions among 16E5, MHC-I and/or other associated proteins. Identical residues are in bold
BPV E5: A Case Study
E5 also plays an important role in cell tumorigenic transformation. One associated cellular target of the bovine papillomavirus BPV-1 E5 is the platelet-derived growth factor β receptor (PDGFβR), a transmembrane receptor tyrosine kinase (Fig. 15.16, left). Under normal circumstances, the ligand PDGF binds to the extracellular domain of its receptor to induce receptor dimerization, leading to the autophosphorylation of tyrosine residues at the receptor intracellular domain, activation of their intrinsic tyrosine kinase activity, and subsequent signal transduction resulting in mitogenesis (Fig. 15.16, middle). While the BPV-1 E5 is not a natural ligand of PDGFβR, it can constitutively activate its receptor tyrosine kinase activity (Fig. 15.16, right) (Drummond-Barbosa et al. 1995) and is therefore an oncoprotein that can lead to host cell transformation.

Model for E5-PDGFβR interaction. Left, monomer of inactive PDGFβR; middle, PDGFβR activation by PDGF binding in the extracellular domain, leading to receptor dimerization, tyrosine phosphorylation in the intracellular domain, and recruitment of cellular signaling substrates (green and purple); right, PDGFβR activation by binding of a BPV-1 E5 dimer to the TMD of the receptor. Horizontal lines represent cell membrane, with the cytoplasm beneath [adapted from DiMaio and Petti (2013)]
Coimmunoprecipitation experiments have shown that the E5-PDGFβR interaction is stable and is important in E5-induced cell transformation (Petti and DiMaio 1992; Goldstein et al. 1992). Deletion of the extracellular ligand-binding domain of PDGFβR did not prevent its cooperation with E5, demonstrating that E5 activation of PDGFβR is ligand independent (Drummond-Barbosa et al. 1995). Also, mutagenesis and chimeric studies of PDGFβR have mapped the binding region to the TMD of the receptor (Cohen et al. 1993; Nappi et al. 2002). The interaction between E5 and PDGFβR is also highly specific, since cooperation was not observed between E5 and PDGFαR, a closely related receptor tyrosine kinase (Goldstein et al. 1994). Molecular dynamics experiments have proposed a model of the E5 dimer, where the Gln17 of monomer 1 and the Asp33 of monomer 2 are on the same face of the dimer that interacts with a molecule of PDGFβR (Surti et al. 1998). NMR studies of E5 peptides in detergent micelles also favor E5 dimerization for complex formation with PDGFβR through its TMD (King et al. 2011).
Concluding Remarks
The interplay between the host cell responses and viral offensive mechanisms yields the final outcome of an infection. For efficient viral replication, the virus must be resourceful in harnessing or disabling the host cellular machinery. In recent years, the role of viroporins as essential players in viral pathogenesis has been established. However, in addition to disrupting cellular ion homeostasis by their channel activity, viroporins also interact with host factors and coordinate with other viral proteins in structural roles. The structural features of these complexes remain poorly understood. Advances in this field will provide useful insights in the design of new antivirals.
References
Adam JL, Briggs MW, McCance DJ (2000) A mutagenic analysis of the E5 protein of human papillomavirus type 16 reveals that E5 binding to the vacuolar H+-ATPase is not sufficient for biological activity, using mammalian and yeast expression systems. Virology 272(2):315–325. https://doi.org/10.1006/viro.2000.0376
Aligeti M, Roder A, Horner SM (2015) Cooperation between the hepatitis C virus p7 and NS5B proteins enhances Virion infectivity. J Virol 89(22):11523–11533. https://doi.org/10.1128/JVI.01185-15
Álvarez E, DeDiego ML, Nieto-Torres JL, Jiménez-Guardeño JM, Marcos-Villar L, Enjuanes L (2010) The envelope protein of severe acute respiratory syndrome coronavirus interacts with the non-structural protein 3 and is ubiquitinated. Virology 402(2):281–291. https://doi.org/10.1016/j.virol.2010.03.015
Anderson KA, Means RL, Huang QH, Kemp BE, Goldstein EG, Selbert MA, Edelman AM, Fremeau RT, Means AR (1999) Components of a calmodulin-dependent protein kinase cascade: molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase β. J Biol Chem 273(48):31880–31889. https://doi.org/10.1074/jbc.273.48.31880
Andresson T, Sparkowski J, Goldstein DJ, Schlegel R (1995) Vacuolar H+-ATPase mutants transform cells and define a binding site for the papillomavirus E5 oncoprotein. J Biol Chem 270(12):6830–6837. https://doi.org/10.1074/jbc.270.12.6830
Anthony SJ, Epstein JH, Murray KA, Navarrete-Macias I, Zambrana-Torrelio CM, Solovyov A, Ojeda-Flores R, Arrigo NC, Islam A, Khan SA, Hosseini P, Bogich TL, Olival KJ, Sanchez-Leon MD, Karesh WB, Goldstein T, Luby SP, Morse SS, Mazet JAK, Daszak P, Lipkin WI (2013) A strategy to estimate unknown viral diversity in mammals. MBio 4(5). ARTN e00598-13. https://doi.org/10.1128/mBio.00598-13
Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R (2008) Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Path 4(3). https://doi.org/10.1371/journal.ppat.1000035
Apps R, Del Prete GQ, Chatterjee P, Lara A, Brumme ZL, Brockman MA, Neil S, Pickering S, Schneider DK, Piechocka-Trocha A, Walker BD, Thomas R, Shaw GM, Hahn BH, Keele BF, Lifson JD, Carrington M (2016) HIV-1 Vpu mediates HLA-C Downregulation. Cell Host and Microbe 19(5):686–695. https://doi.org/10.1016/j.chom.2016.04.005
Ashrafi GH, Haghshenas M, Marchetti B, Campo MS (2006) E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int J Cancer 119(9):2105–2112. https://doi.org/10.1002/ijc.22089
Ashrafi GH, Haghshenas MR, Marchetti B, O'Brien PM, Campo MS (2005) E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int J Cancer 113(2):276–283. https://doi.org/10.1002/ijc.20558
Au KS, Chan WK, Burns JW, Estes MK (1989) Receptor activity of rotavirus nonstructural glycoprotein NS28. J Virol 63(11):4553–4562
Au KS, Mattion NM, Estes MK (1993) A subviral particle binding domain on the rotavirus nonstructural glycoprotein NS28. Virology 194(2):665–673. https://doi.org/10.1006/viro.1993.1306
Ball JM, Schroeder ME, Williams CV, Schroeder F, Parr RD (2013) Mutational analysis of the rotavirus NSP4 enterotoxic domain that binds to caveolin-1. Virol J 10. https://doi.org/10.1186/1743-422X-10-336
Ball JM, Tian P, Zeng CQY, Morris AP, Estes MK (1996) Age-dependent diarrhea induced by a rotaviral nonstructural glycoprotein. Science 272(5258):101–104
Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, Donovan RS, Shinjo F, Liu Y, Dembowy J, Taylor IW, Luga V, Przulj N, Robinson M, Suzuki H, Hayashizaki Y, Jurisica I, Wrana JL (2005) High-throughput mapping of a dynamic signaling network in mammalian cells. Science 307(5715):1621–1625. https://doi.org/10.1126/science.1105776
Beale R, Wise H, Stuart A, Ravenhill BJ, Digard P, Randow F (2014) A LC3-interacting motif in the influenza a virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host and Microbe 15(2):239–247. https://doi.org/10.1016/j.chom.2014.01.006
Berkova Z, Crawford SE, Trugnan G, Yoshimori T, Morris AP, Estes MK (2006) Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J Virol 80(12):6061–6071. https://doi.org/10.1128/JVI.02167-05
Binette J, Dubé M, Mercier J, Halawani D, Latterich M, Cohen EA (2007) Requirements for the selective degradation of CD4 receptor molecules by the human immunodeficiency virus type 1 Vpu protein in the endoplasmic reticulum. Retrovirology 4. https://doi.org/10.1186/1742-4690-4-75
Boshuizen JA, Rossen JWA, Sitaram CK, Kimenai FFP, Simons-Oosterhuis Y, Laffeber C, Büller HA, Einerhand AWC (2004) Rotavirus enterotoxin NSP4 binds to the extracellular matrix proteins laminin-β3 and fibronectin. J Virol 78(18):10045–10053. https://doi.org/10.1128/JVI.78.18.10045-10053.2004
Bour S, Schubert U, Strebel K (1995) The human immunodeficiency virus type 1 Vpu protein specifically binds to the cytoplasmic domain of CD4: implications for the mechanism of degradation. J Virol 69(3):1510–1520
Bourmakina SV, García-Sastre A (2003) Reverse genetics studies on the filamentous morphology of influenza a virus. J Gen Virol 84(3):517–527. https://doi.org/10.1099/vir.0.18803-0
Bowman GD, Nodelman IM, Levy O, Lin SL, Tian P, Zamb TJ, Udem SA, Venkataraghavan B, Schutt CE (2000) Crystal structure of the oligomerization domain of NSP4 from rotavirus reveals a core metal-binding site. J Mol Biol 304(5):861–871. https://doi.org/10.1006/jmbi.2000.4250
Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC (2003) Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 160(7):1115–1127. https://doi.org/10.1083/jcb.200212059
Bugarc̀ić A, Taylor JA (2006) Rotavirus nonstructural glycoprotein NSP4 is secreted from the apical surfaces of polarized epithelial cells. J Virol 80(24):12343–12349. https://doi.org/10.1128/JVI.01378-06
Bukreyev A, Whitehead SS, Murphy BR, Collins PL (1997) Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site-specific attenuation in the respiratory tract of the mouse. J Virol 71(12):8973–8982
Carrasco L (1995) Modification of membrane permeability by animal viruses. Adv Virus Res 45:61–112
Chacko AR, Arifullah M, Sastri NP, Jeyakanthan J, Ueno G, Sekar K, Read RJ, Dodson EJ, Rao DC, Suguna K (2011) Novel pentameric structure of the diarrhea-inducing region of the rotavirus enterotoxigenic protein NSP4. J Virol 85(23):12721–12732. https://doi.org/10.1128/JVI.00349-11
Champeimont R, Laine E, Hu SW, Penin F, Carbone A (2016) Coevolution analysis of hepatitis C virus genome to identify the structural and functional dependency network of viral proteins. Sci Rep 6. https://doi.org/10.1038/srep26401
Chen BJ, Leser GP, Jackson D, Lamb RA (2008) The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J Virol 82(20):10059–10070
Chen SL, Lin ST, Tsai TC, Hsiao WC, Tsao YP (2007) ErbB4 (JM-b/CYT-1)-induced expression and phosphorylation of c-Jun is abrogated by human papillomavirus type 16 E5 protein. Oncogene 26(1):42–53. https://doi.org/10.1038/sj.onc.1209768
Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O'Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW (2001) A novel influenza a virus mitochondrial protein that induces cell death. Nat Med 7(12):1306–1312. https://doi.org/10.1038/nm1201-1306
Cohen BD, Goldstein DJ, Rutledge L, Vass WC, Lowy DR, Schlegel R, Schiller JT (1993) Transformation-specific interaction of the bovine papillomavirus E5 oncoprotein with the platelet-derived growth factor receptor transmembrane domain and the epidermal growth factor receptor cytoplasmic domain. J Virol 67(9):5303–5311
Cohen EA, Terwilliger EF, Sodroski JG, Haseltine WA (1988) Identification of a protein encoded by the vpu gene of HIV-1. Nature 334(6182):532–534
Collins PL, Huang YT, Wertz GW (1984) Identification of a tenth mRNA of respiratory syncytial virus and assignment of polypeptides to the 10 viral genes. J Virol 49(2):572–578
Collins PL, Mottet G (1993) Membrane orientation and oligomerization of the small hydrophobic protein of human respiratory syncytial virus. J Gen Virol 74:1445–1450
Collins PL, Olmsted RA, Johnson PR (1990) The small hydrophobic protein of human respiratory syncytial virus: comparison between antigenic subgroups a and B. J Gen Virol 71(Pt 7):1571–1576
Conrad M, Bubb VJ, Schlegel R (1993) The human papillomavirus type 6 and 16 E5 proteins are membrane-associated proteins which associate with the 16-kilodalton pore-forming protein. J Virol 67(10):6170–6178
Cook GA, Dawson LA, Tian Y, Opella SJ (2013) Three-dimensional structure and interaction studies of hepatitis C virus p7 in 1,2-dihexanoyl- sn -glycero-3-phosphocholine by solution nuclear magnetic resonance. Biochemistry 52(31):5295–5303. https://doi.org/10.1021/bi4006623
Cook GA, Zhang H, Park SH, Wang Y, Opella SJ (2010) Comparative NMR studies demonstrate profound differences between two viroporins: p7 of HCV and Vpu of HIV-1. Biochim Biophys Acta 1808 (2):554–560. https://doi.org/S0005-2736(10)00286-5 [pii] https://doi.org/10.1016/j.bbamem.2010.08.005
Coombs KM (2006) Reovirus structure and morphogenesis. Current Topics in Microbiology and Immunology, vol 309
Coric P, Saribas AS, Abou-Gharbia M, Childers W, Condra JH, White MK, Safak M, Bouaziz S (2017) Nuclear magnetic resonance structure of the human Polyoma JC virus Agnoprotein. J Cell Biochem. https://doi.org/10.1002/jcb.25977
Coric P, Saribas AS, Abou-Gharbia M, Childers W, White MK, Bouaziz S, Safak M (2014) Nuclear magnetic resonance structure revealed that the human polyomavirus JC virus agnoprotein contains an α-helix encompassing the Leu/Ile/Phe-rich domain. J Virol 88(12):6556–6575. https://doi.org/10.1128/JVI.00146-14
Corse E, Machamer CE (2003) The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 312(1):25–34
Cortese MS, Ashrafi GH, Campo MS (2010) All 4 di-leucine motifs in the first hydrophobic domain of the E5 oncoprotein of human papillomavirus type 16 are essential for surface MHC class I downregulation activity and E5 endomembrane localization. Int J Cancer 126(7):1675–1682. https://doi.org/10.1002/ijc.25004
Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP (1997) Identification of peptide and protein ligands for the caveolin- scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem 272(10):6525–6533. https://doi.org/10.1074/jbc.272.10.6525
Crawford SE, Hyser JM, Utama B, Estes MK (2012) Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-β signaling is required for rotavirus replication. Proc Natl Acad Sci U S A 109(50):E3405–E3413. https://doi.org/10.1073/pnas.1216539109
Darbinyan A, Darbinian N, Safak M, Radhakrishnan S, Giordano A, Khalili K (2002) Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene 21(36):5574–5581. https://doi.org/10.1038/sj.onc.1205744
Darbinyan A, Siddiqui KM, Slonina D, Darbinian N, Amini S, White MK, Khalili K (2004) Role of JC virus agnoprotein in DNA repair. J Virol 78(16):8593–8600. https://doi.org/10.1128/JVI.78.16.8593-8600.2004
de Chassey B, Meyniel-Schicklin L, Vonderscher J, André P, Lotteau V (2014) Virus-host interactomics: new insights and opportunities for antiviral drug discovery. Genome Med 6(11). https://doi.org/10.1186/s13073-014-0115-1
De Chassey B, Navratil V, Tafforeau L, Hiet MS, Aublin-Gex A, Agaugué S, Meiffren G, Pradezynski F, Faria BF, Chantier T, Le Breton M, Pellet J, Davoust N, Mangeot PE, Chaboud A, Penin F, Jacob Y, Vidalain PO, Vidal M, André P, Rabourdin-Combe C, Lotteau V (2008) Hepatitis C virus infection protein network. Mol Syst Biol 4. https://doi.org/10.1038/msb.2008.66
DeDiego ML, Álvarez E, Almazán F, Rejas MT, Lamirande E, Roberts A, Shieh WJ, Zaki SR, Subbarao K, Enjuanes L (2007) A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J Virol 81(4):1701–1713. https://doi.org/10.1128/JVI.01467-06
DeDiego ML, Nieto-Torres JL, Jimenez-Guardeno JM, Regla-Nava JA, Alvarez E, Oliveros JC, Zhao J, Fett C, Perlman S, Enjuanes L (2011) Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Path 7(10):e1002315. https://doi.org/10.1371/journal.ppat.1002315
DeDiego ML, Pewe L, Alvarez E, Rejas MT, Perlman S, Enjuanes L (2008) Pathogenicity of severe acute respiratory coronavirus deletion mutants in hACE-2 transgenic mice. Virology 376(2):379–389. https://doi.org/10.1016/j.virol.2008.03.005
Deyde VM, Xu X, Bright RA, Shaw M, Smith CB, Zhang Y, Shu Y, Gubareva LV, Cox NJ, Klimov AI (2007) Surveillance of resistance to adamantanes among influenza a(H3N2) and a(H1N1) viruses isolated worldwide. J Infect Dis 196(2):249–257
DiMaio D, Petti LM (2013) The E5 proteins. Virology 445(1–2):99–114. https://doi.org/10.1016/j.virol.2013.05.006
Do HQ, Wittlich M, Glück JM, Möckel L, Willbold D, Koenig BW, Heise H (2013) Full-length Vpu and human CD4(372-433) in phospholipid bilayers as seen by magic angle spinning NMR. Biol Chem 394(11):1453–1463. https://doi.org/10.1515/hsz-2013-0194
Doms RW, Trono D (2000) The plasma membrane as a combat zone in the HIV battlefield. Genes Dev 14(21):2677–2688. https://doi.org/10.1101/gad.833300
Dotson D, Woodruff EA, Villalta F, Dong X (2016) Filamin a is involved in HIV-1 Vpu-mediated evasion of host restriction by modulating tetherin expression. J Biol Chem 291(8):4236–4246. https://doi.org/10.1074/jbc.M115.708123
Dowell SF, Anderson LJ, Gary HE Jr, Erdman DD, Plouffe JF, File TM Jr, Marston BJ, Breiman RF (1996) Respiratory syncytial virus is an important cause of community-acquired lower respiratory infection among hospitalized adults. J Infect Dis 174(3):456–462
Drummond-Barbosa D, Vaillancourt RR, Kazlauskas A, Dimaio D (1995) Ligand-independent activation of the platelet-derived growth factor β receptor: requirements for bovine papillomavirus E5-induced mitogenic signaling. Mol Cell Biol 15(5):2570–2581
Dubé M, Bego MG, Paquay C, Cohen TA (2010) Modulation of HIV-1-host interaction: role of the Vpu accessory protein. Retrovirology 7. https://doi.org/10.1186/1742-4690-7-114
Dubé M, Paquay C, Roy BB, Bego MG, Mercier J, Cohen EA (2011) HIV-1 Vpu antagonizes BST-2 by interfering mainly with the trafficking of newly synthesized BST-2 to the cell surface. Traffic 12(12):1714–1729. https://doi.org/10.1111/j.1600-0854.2011.01277.x
Elliott EI, Sutterwala FS (2015) Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev 265(1):35–52. https://doi.org/10.1111/imr.12286
Endo S, Okada Y, Orba Y, Nishihara H, Tanaka S, Nagashima K, Sawa H (2003) JC virus agnoprotein colocalizes with tubulin. J Neurovirol 9(SUPPL. 1):10–14
Enjuanes L, Brian D, Cavanagh D, Holmes K, Lai MMC, Laude H, Masters P, Rottier P, Siddell SG, Spaan WJM, Taguchi F, Talbot P (2000) Coronaviridae. In: van MHV R, Fauquet CM, Bishop DHL et al (eds) Virus taxonomy. Classification and nomenclature of viruses. Academic Press, San Diego, pp 835–849
Enjuanes L, Gorbalenya AE, de Groot RJ, Cowley JA, Ziebuhr J, Snijder EJ (2008) The Nidovirales. Encyclopedia of Virology:419–430
Estes MK, Greenberg HB (2013) Rotaviruses. Fields Virology:1347–1401
Fan Y, Mok CKP, Chan MCW, Zhang Y, Nal B, Kien F, Bruzzone R, Sanyal S (2017) Cell cycle-independent role of Cyclin D3 in host restriction of influenza virus infection. J Biol Chem 292(12):5070–5088. https://doi.org/10.1074/jbc.M117.776112
Fields BN, Knipe DM, Howley PM (2013) Fields virology. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia
Fischer WB, Li LH, Mahato DR, Wang YT, Chen CP (2014) Viral channel proteins in intracellular protein-protein communication: Vpu of HIV-1, E5 of HPV16 and p7 of HCV. Biochim Biophys Acta 1838(4):1113–1121. https://doi.org/10.1016/j.bbamem.2013.08.017
Forni D, Cagliani R, Clerici M, Sironi M (2017) Molecular evolution of human coronavirus genomes. Trends Microbiol 25(1):35–48. https://doi.org/10.1016/j.tim.2016.09.001
Fuentes S, Tran KC, Luthra P, Teng MN, He B (2007) Function of the respiratory syncytial virus small hydrophobic protein. J Virol 81(15):8361–8366
Gan SW, Ng L, Xin L, Gong X, Torres J (2008) Structure and ion channel activity of the human respiratory syncytial virus (hRSV) small hydrophobic protein transmembrane domain. Protein Sci 17:813–820
Gan SW, Surya W, Vararattanavech A, Torres J (2014) Two different conformations in hepatitis C virus p7 protein account for proton transport and dye release. PLoS One 9(1):e78494. https://doi.org/10.1371/journal.pone.0078494
Gan SW, Tan E, Lin X, Yu D, Wang J, Tan GM-Y, Vararattanavech A, Yeo CY, Soon CH, Soong TW, Pervushin K, Torres J (2012) The small hydrophobic protein of the human respiratory syncytial virus forms pentameric ion channels. J Biol Chem 287(29):24671–24689
Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, Lee M, Strowig T, Arrey F, Conenello G, Pypaert M, Andersen J, García-Sastre A, Münz C (2009) Matrix protein 2 of influenza a virus blocks Autophagosome fusion with lysosomes. Cell Host and Microbe 6(4):367–380. https://doi.org/10.1016/j.chom.2009.09.005
Gerber D, Maerkl SJ, Quake SR (2009) An in vitro microfluidic approach to generating protein-interaction networks. Nat Methods 6(1):71–74. https://doi.org/10.1038/nmeth.1289
Germain MA, Chatel-Chaix L, Gagné B, Bonneil E, Thibault P, Pradezynski F, De Chassey B, Meyniel-Schicklin L, Lotteau V, Baril M, Lamarre D (2014) Elucidating novel hepatitis C virus-host interactions using combined mass spectrometry and functional genomics approaches. Mol Cell Proteomics 13(1):184–203. https://doi.org/10.1074/mcp.M113.033803
Gieswein CE, Sharom FJ, Wildeman AG (2003) Oligomerization of the E5 protein of human papillomavirus type 16 occurs through multiple hydrophobic regions. Virology 313(2):415–426. https://doi.org/10.1016/S0042-6822(03)00296-4
Goldstein DJ, Andresson T, Sparkowski JJ, Schlegel R (1992) The BPV-1 E5 protein, the 16 kDa membrane pore-forming protein and the PDGF receptor exist in a complex that is dependent on hydrophobic transmembrane interactions. EMBO J 11(13):4851–4859
Goldstein DJ, Fmbow ME, Andresson T, McLean P, Smith K, Bubb V, Schlegel R (1991) Bovine papillomavirus E5 oncoprotein binds to the 16K component of vacuolar H+-ATPases. Nature 352(6333):347–349
Goldstein DJ, Li W, Wang LM, Heidaran MA, Aaronson S, Shinn R, Schlegel R, Pierce JH (1994) The bovine papillomavirus type 1 E5 transforming protein specifically binds and activates the β-type receptor for the platelet-derived growth factor but not other related tyrosine kinase-containing receptors to induce cellular transformation. J Virol 68(7):4432–4441
Griffin SDC, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MPG, Rowlands DJ, Klenk HD (2003) The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, amantadine. FEBS Lett 535(1–3):34–38
Gruener M, Bravo IG, Momburg F, Alonso A, Tomakidi P (2007) The E5 protein of the human papillomavirus type 16 down-regulates HLA-I surface expression in calnexin-expressing but not in calnexin-deficient cells. Virol J 4. https://doi.org/10.1186/1743-422X-4-116
Gu RX, Liu LA, Wei DQ (2013) Structural and energetic analysis of drug inhibition of the influenza a M2 proton channel. Trends Pharmacol Sci 34(10):571–580. https://doi.org/10.1016/j.tips.2013.08.003
Guan Z, Liu D, Mi S, Zhang J, Ye Q, Wang M, Gao GF, Yan J (2010) Interaction of Hsp40 with influenza virus M2 protein: implications for PKR signaling pathway. Protein and Cell 1(10):944–955. https://doi.org/10.1007/s13238-010-0115-x
Hagai T, Azia A, Babu MM, Andino R (2014) Use of host-like peptide motifs in viral proteins is a prevalent strategy in host-virus interactions. Cell Rep 7:1729–1739
Hagen N, Bayers K, Ros̈ch K, Schindler M (2014) The intraviral protein interaction network of hepatitis C virus. Mol Cell Proteomics 13(7):1676–1689. https://doi.org/10.1074/mcp.M113.036301
Hall CB, Walsh EE, Schnabel KC, Long CE, Mcconnochie KM, Hildreth SW, Anderson LJ (1990) Occurrence of group-a and group-B of respiratory syncytial virus over 15 years - associated epidemiologic and clinical characteristics in hospitalized and ambulatory children. J Infect Dis 162(6):1283–1290
Haller C, Müller B, Fritz JV, Lamas-Murua M, Stolp B, Pujol FM, Keppler OT, Fackler OT (2014) HIV-1 Nef and Vpu are functionally redundant broad-spectrum modulators of cell surface receptors, including tetraspanins. J Virol 88(24):14241–14257. https://doi.org/10.1128/JVI.02333-14
Harris BZ, Lim WA (2001) Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci 114(18):3219–3231
Hauser H, Lopez LA, Yang SJ, Oldenburg JE, Exline CM, Guatelli JC, Cannon PM (2010) HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 7. https://doi.org/10.1186/1742-4690-7-51
Hay AJ, Gregory V, Douglas AR, Yi PL (2001) The evolution of human influenza viruses. Philosophical Transactions of the Royal Society B: Biological Sciences 356(1416):1861–1870. https://doi.org/10.1098/rstb.2001.0999
Hayden FG, De Jong MD (2011) Emerging influenza antiviral resistance threats. J Infect Dis 203(1):6–10. https://doi.org/10.1093/infdis/jiq012
Helenius A (1992) Unpacking the incoming influenza virus. Cell 69(4):577–578. https://doi.org/10.1016/0092-8674(92)90219-3
Henkel M, Mitzner D, Henklein P, Meyer-Almes FJ, Moroni A, DiFrancesco ML, Henkes LM, Kreim M, Kast SM, Schubert U, Thiel G (2010) Proapoptotic influenza a virus protein PB1-F2 forms a nonselective ion channel. PLoS One 5(6). https://doi.org/10.1371/journal.pone.0011112
Ho Y, Lin PH, Liu CYY, Lee SP, Chao YC (2004) Assembly of human severe acute respiratory syndrome coronavirus-like particles. Biochem Biophys Res Commun 318(4):833–838. https://doi.org/10.1016/j.bbrc.2004.04.111
Hogue BG, Machamer CE (2008) Coronavirus structural proteins and virus assembly. In: Perlman S, Gallagher T, Snijder EJ (eds) Nidoviruses, Washington DC, pp 179–200
Holmes KV (2003) SARS coronavirus: a new challenge for prevention and therapy. J Clin Invest 111(11):1605–1609. https://doi.org/10.1172/JCI18819
Hsu K, Seharaseyon J, Dong P, Bour S, Marbán E (2004) Mutual functional destruction of HIV-1 Vpu and host TASK-1 channel. Mol Cell 14(2):259–267. https://doi.org/10.1016/S1097-2765(04)00183-2
Hu S, Yin L, Mei S, Li J, Xu F, Sun H, Liu X, Cen S, Liang C, Li A, Guo F (2017) BST-2 restricts IAV release and is countered by the viral M2 protein. Biochem J 474(5):715–730. https://doi.org/10.1042/BCJ20160861
Hwang ES, Nottoli T, Dimaio D (1995) The HPV16 E5 protein: expression, detection, and stable complex formation with transmembrane proteins in COS cells. Virology 211(1):227–233. https://doi.org/10.1006/viro.1995.1395
Hyser JM, Collinson-Pautz MR, Utama B, Estes MK (2010) Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. MBio 1(5). https://doi.org/10.1128/mBio.00265-10
Hyser JM, Estes MK (2015) Pathophysiological consequences of calcium-conducting Viroporins. Annual Review of Virology 2:473–496. https://doi.org/10.1146/annurev-virology-100114-054846
Hyser JM, Utama B, Crawford SE, Broughman JR, Estes MK (2013) Activation of the endoplasmic reticulum calcium sensor STIM1 and store-operated calcium entry by rotavirus requires NSP4 viroporin activity. J Virol 87(24):13579–13588. https://doi.org/10.1128/JVI.02629-13
Ichinohe T, Pang IK, Iwasaki A (2010) Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11(5):404–410. https://doi.org/10.1038/ni.1861
Imbert I, Snijder EJ, Dimitrova M, Guillemot JC, Lécine P, Canard B (2008) The SARS-coronavirus PLnc domain of nsp3 as a replication/transcription scaffolding protein. Virus Res 133(2):136–148. https://doi.org/10.1016/j.virusres.2007.11.017
Ito M, Yanagi Y, Ichinohe T (2012) Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Path 8(8):e1002857. https://doi.org/10.1371/journal.ppat.1002857
Ito N, Mossel EC, Narayanan K, Popov VL, Huang C, Inoue T, Peters CJ, Makino S (2005) Severe acute respiratory syndrome coronavirus 3a protein is a viral structural protein. J Virol 79(5):3182–3186. https://doi.org/10.1128/JVI.79.5.3182-3186.2005
Javier RT, Rice AP (2011) Emerging theme: cellular PDZ proteins as common targets of pathogenic viruses. J Virol 85(22):11544–11556. https://doi.org/10.1128/JVI.05410-11
Ji HL, Song W, Gao Z, Su XF, Nie HG, Jiang Y, Peng JB, He YX, Liao Y, Zhou YJ, Tousson A, Matalon S (2009) SARS-CoV proteins decrease levels and activity of human ENaC via activation of distinct PKC isoforms. Am J Physiol Lung Cell Mol Physiol 296(3):L372–L383. https://doi.org/10.1152/ajplung.90437.2008
Jia X, Weber E, Tokarev A, Lewinski M, Rizk M, Suarez M, Guatelli J, Xiong Y (2014) Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. elife 2014(3). https://doi.org/10.7554/eLife.02362
Jimenez-Guardeño JM, Nieto-Torres JL, DeDiego ML, Regla-Nava JA, Fernandez-Delgado R, Castaño-Rodriguez C, Enjuanes L (2014) The PDZ-binding motif of severe acute respiratory syndrome coronavirus envelope protein is a determinant of viral pathogenesis. PLoS Path 10(8). https://doi.org/10.1371/journal.ppat.1004320
Jirasko V, Montserret R, Appel N, Janvier A, Eustachi L, Brohm C, Steinmann E, Pietschmann T, Penin F, Bartenschlagerd R (2008) Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J Biol Chem 283(42):28546–28562. https://doi.org/10.1074/jbc.M803981200
Jirasko V, Montserret R, Lee JY, Gouttenoire J, Moradpour D, Penin F, Bartenschlager R (2010) Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Path 6(12). https://doi.org/10.1371/journal.ppat.1001233
Kabsch K, Mossadegh N, Kohl A, Komposch G, Schenkel J, Alonso A, Tomakidi P (2004) The HPV-16 E5 protein inhibits TRAIL- and FasL-mediated apoptosis in human keratinocyte raft cultures. Intervirology 47(1):48–56. https://doi.org/10.1159/000076642
Kaniowska D, Kaminski R, Amini S, Radhakrishnan S, Rappaport J, Johnson E, Khalili K, Del Valle L, Darbinyan A (2006) Cross-interaction between JC virus agnoprotein and human immunodeficiency virus type 1 (HIV-1) tat modulates transcription of the HIV-1 long terminal repeat in glial cells. J Virol 80(18):9288–9299. https://doi.org/10.1128/JVI.02138-05
Kanzawa N, Nishigaki K, Hayashi T, Ishii Y, Furukawa S, Niiro A, Yasui F, Kohara M, Morita K, Matsushima K, Le MQ, Masuda T, Kannagi M (2006) Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-κB activation. FEBS Lett 580(30):6807–6812. https://doi.org/10.1016/j.febslet.2006.11.046
Kargel A, Schmidt U, Buchholz UJ (2001) Recombinant bovine respiratory syncytial virus with deletions of the G or SH genes: G and F proteins binds heparin. J Gen Virol 82(3):631–640
Karron RA, Wright PF, Belshe RB, Thumar B, Casey R, Newman F, Polack FP, Randolph VB, Deatly A, Hackell J, Gruber W, Murphy BR, Collins PL (2005) Identification of a recombinant live attenuated respiratory syncytial virus vaccine candidate that is highly attenuated in infants. J Infect Dis 191(7):1093–1104. https://doi.org/10.1086/427813
Kell B, Jewers RJ, Cason J, Pakarian F, Kaye JN, Best JM (1994) Detection of E5 oncoprotein in human papillomavirus type 16-positive cervical scrapes using antibodies raised to synthetic peptides. J Gen Virol 75(9):2451–2456
Kerkau T, Bacik I, Bennink JR, Yewdell JW, Hünig T, Schimpl A, Schubert U (1997) The human immunodeficiency virus type 1 (HIV-1)vpu protein interferes with an early step in the biosynthesis of major histocompatibility complex (MHC) class I molecules. J Exp Med 185(7):1295–1305. https://doi.org/10.1084/jem.185.7.1295
King G, Oates J, Patel D, Van Den Berg HA, Dixon AM (2011) Towards a structural understanding of the smallest known oncoprotein: investigation of the bovine papillomavirus E5 protein using solution-state NMR. Biochim Biophys Acta Biomembr 1808(6):1493–1501. https://doi.org/10.1016/j.bbamem.2010.11.004
Kipper S, Hamad S, Caly L, Avrahami D, Bacharach E, Jans DA, Gerber D, Bajorek M (2015) New host factors important for respiratory syncytial virus replication revealed by a novel microfluidics screen for interactors of matrix protein. Mol Cell Proteomics
Klimkait T, Strebel K, Hoggan MD, Martin MA, Orenstein JM (1990) The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J Virol 64(2):621–629
Kobayashi T, Ode H, Yoshida T, Sato K, Gee P, Yamamoto SP, Ebina H, Strebel K, Sato H, Koyanagi Y (2011) Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J Virol 85(2):932–945. https://doi.org/10.1128/JVI.01668-10
Kotnik Halavaty K, Regan J, Mehta K, Laimins L (2014) Human papillomavirus E5 oncoproteins bind the A4 endoplasmic reticulum protein to regulate proliferative ability upon differentiation. Virology 452-453:223–230. https://doi.org/10.1016/j.virol.2014.01.013
Krawczyk E, Hanover JA, Schlegel R, Suprynowicz FA (2008) Karyopherin β3: a new cellular target for the HPV-16 E5 oncoprotein. Biochem Biophys Res Commun 371(4):684–688. https://doi.org/10.1016/j.bbrc.2008.04.122
Krawczyk E, Suprynowicz FA, Hebert JD, Kamonjoh CM, Schlegel R (2011) The human papillomavirus type 16 E5 oncoprotein translocates calpactin I to the perinuclear region. J Virol 85(21):10968–10975. https://doi.org/10.1128/JVI.00706-11
Krawczyk E, Suprynowicz FA, Sudarshan SR, Schlegel R (2010) Membrane orientation of the human papillomavirus type 16 E5 oncoprotein. J Virol 84(4):1696–1703. https://doi.org/10.1128/JVI.01968-09
Kueck T, Foster TL, Weinelt J, Sumner JC, Pickering S, Neil SJD (2015) Serine phosphorylation of HIV-1 Vpu and its binding to Tetherin regulates interaction with Clathrin adaptors. PLoS Path 11(8). https://doi.org/10.1371/journal.ppat.1005141
Kueck T, Neil SJD (2012) A cytoplasmic tail determinant in HIV-1 vpu mediates targeting of tetherin for endosomal degradation and counteracts interferon-induced restriction. PLoS Path 8(3). https://doi.org/10.1371/journal.ppat.1002609
Kuo L, Masters PS (2010) Evolved variants of the membrane protein can partially replace the envelope protein in murine coronavirus assembly. J Virol 84(24):12872–12885. https://doi.org/10.1128/JVI.01850-10
Ladasky JJ, Boyle S, Seth M, Li H, Pentcheva T, Abe F, Steinberg SJ, Edidin M (2006) Bap31 enhances the endoplasmic reticulum export and quality control of human class I MHC molecules. J Immunol 177(9):6172–6181
Lamb RA, Choppin PW (1983) The gene structure and replication of influenza virus. Annu Rev Biochem 52:467–506
Lambelé M, Koppensteiner H, Symeonides M, Roy NH, Chan J, Schindler M, Thali M (2015) Vpu is the main determinant for tetraspanin downregulation in HIV-1-infected cells. J Virol 89(6):3247–3255. https://doi.org/10.1128/JVI.03719-14
Lau D, Kwan W, Guatelli J (2011) Role of the endocytic pathway in the counteraction of BST-2 by human lentiviral pathogens. J Virol 85(19):9834–9846. https://doi.org/10.1128/JVI.02633-10
Lazarczyk M, Pons C, Mendoza JA, Cassonnet P, Jacob Y, Favre M (2008) Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. J Exp Med 205(1):35–42. https://doi.org/10.1084/jem.20071311
le Tortorec A, Willey S, Neil SJD (2011) Antiviral inhibition of enveloped virus release by Tetherin/BST-2: action and counteraction. Virus 3(5):520–540. https://doi.org/10.3390/v3050520
Lewinski MK, Jafari M, Zhang H, Opella SJ, Guatelli J (2015) Membrane anchoring by a C-terminal tryptophan enables HIV-1 Vpu to displace bone marrow stromal antigen 2 (BST2) from sites of viral assembly. J Biol Chem 290(17):10919–10933. https://doi.org/10.1074/jbc.M114.630095
Li Y, Jain N, Limpanawat S, To J, Quistgaard EM, Nordlund P, Thanabalu T, Torres J (2015) Interaction between human BAP31 and respiratory syncytial virus small hydrophobic (SH) protein. Virology 482:105–110. https://doi.org/10.1016/j.virol.2015.03.034
Li Y, Surya W, Claudine S, Torres J (2014a) Structure of a conserved Golgi complex-targeting signal in coronavirus envelope proteins. J Biol Chem 289(18):12535–12549. https://doi.org/10.1074/jbc.M114.560094
Li Y, To J, Verdia-Baguena C, Dossena S, Surya W, Huang M, Paulmichl M, Liu DX, Aguilella VM, Torresa J (2014b) Inhibition of the human respiratory syncytial virus small hydrophobic protein and structural variations in a Bicelle environment. J Virol 89(20):11899–11914. https://doi.org/10.1128/Jvi.00839-14
Lim KP, Liu DX (2001) The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre-Golgi compartments and physical interaction between the envelope and membrane proteins. J Biol Chem 276(20):17515–17523
Lin C, Lindenbach BD, Prágai BM, McCourt DW, Rice CM (1994) Processing in the hepatitis C virus E2-NS2 region: identification of p7 and two distinct E2-specific products with different C termini. J Virol 68(8):5063–5073
Lindenbach BD, Rice CM (2013) The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol 11(10):688–700. https://doi.org/10.1038/nrmicro3098
Lindner HA, Fotouhi-Ardakani N, Lytvyn V, Lachance P, Sulea T, Ménard R (2005) The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol 79(24):15199–15208. https://doi.org/10.1128/JVI.79.24.15199-15208.2005
Lou Z, Sun Y, Rao Z (2014) Current progress in antiviral strategies. Trends Pharmacol Sci 35(2):86–102. https://doi.org/10.1016/j.tips.2013.11.006
Low KW, Tan T, Ng K, Tan BH, Sugrue RJ (2008) The RSV F and G glycoproteins interact to form a complex on the surface of infected cells. Biochem Biophys Res Commun 366(2):308–313. https://doi.org/10.1016/j.bbrc.2007.11.042
Lu W, Zheng BJ, Xu K, Schwarz W, Du LY, Wong CKL, Chen JD, Duan SM, Deubel V, Sun B (2006) Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc Natl Acad Sci U S A 103(33):12540–12545. https://doi.org/10.1073/pnas.0605402103
Luik P, Chew C, Aittoniemi J, Chang J, Wentworth P, Jr., Dwek RA, Biggin PC, Venien-Bryan C, Zitzmann N (2009) The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc Natl Acad Sci U S A 106 (31):12712–12716. https://doi.org/0905966106 [pii] doi:https://doi.org/10.1073/pnas.0905966106
Lukhele S, Cohen A (2017) Conserved residues within the HIV-1 Vpu transmembrane-proximal hinge region modulate BST2 binding and antagonism. Retrovirology 14(1). https://doi.org/10.1186/s12977-017-0345-6
Ma H, Kien F, Manière M, Zhang Y, Lagarde N, Tse KS, Poon LLM, Nal B (2012) Human annexin A6 interacts with influenza a virus protein M2 and negatively modulates infection. J Virol 86(3):1789–1801. https://doi.org/10.1128/JVI.06003-11
Ma Y, Anantpadma M, Timpe JM, Shanmugam S, Singh SM, Lemon SM, Yi M (2011) Hepatitis C virus NS2 protein serves as a scaffold for virus assembly by interacting with both structural and nonstructural proteins. J Virol 85(1):86–97. https://doi.org/10.1128/JVI.01070-10
Madjo U, Leymarie O, Frémont S, Kuster A, Nehlich M, Gallois-Montbrun S, Janvier K, Berlioz-Torrent C (2016) LC3C contributes to Vpu-mediated antagonism of BST2/Tetherin restriction on HIV-1 release through a non-canonical autophagy pathway. Cell Rep 17(9):2221–2233. https://doi.org/10.1016/j.celrep.2016.10.045
Maeda J, Maeda A, Makino S (1999) Release of coronavirus E protein in membrane vesicles from virus- infected cells and E protein-expressing cells. Virology 263(2):265–272. https://doi.org/10.1006/viro.1999.9955
Magadán JG, Bonifacino JS (2012) Transmembrane domain determinants of CD4 downregulation by HIV-1 Vpu. J Virol 86(2):757–772. https://doi.org/10.1128/jvi.05933-11
Magadán JG, Pérez-Victoria FJ, Sougrat R, Ye Y, Strebel K, Bonifacino JS (2010) Multilayered mechanism of CD4 downregulation by HIV-1 vpu involving distinct ER retention and ERAD targeting steps. PLoS Path 6(4):1–18
Mangeat B, Cavagliotti L, Lehmann M, Gers-Huber G, Kaur I, Thomas Y, Kaiser L, Piguet V (2012) Influenza virus partially counteracts restriction imposed by tetherin/BST-2. J Biol Chem 287(26):22015–22029. https://doi.org/10.1074/jbc.M111.319996
Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R (1998) A novel human WD protein, h-βTrCP, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell 1(4):565–574
Matheson NJ, Sumner J, Wals K, Rapiteanu R, Weekes MP, Vigan R, Weinelt J, Schindler M, Antrobus R, Costa ASH, Frezza C, Clish CB, Neil SJD, Lehner PJ (2015) Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host and Microbe 18(4):409–423. https://doi.org/10.1016/j.chom.2015.09.003
Matusali G, Potestá M, Santoni A, Cerboni C, Doria M (2012) The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell-activating ligand PVR. J Virol 86(8):4496–4504. https://doi.org/10.1128/JVI.05788-11
McAuley JL, Tate MD, MacKenzie-Kludas CJ, Pinar A, Zeng W, Stutz A, Latz E, Brown LE, Mansell A (2013) Activation of the NLRP3 Inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Path 9(5). https://doi.org/10.1371/journal.ppat.1003392
McCown MF, Pekosz A (2006) Distinct domains of the influenza a virus M2 protein cytoplasmic tail mediate binding to the M1 protein and facilitate infectious virus production. J Virol 80(16):8178–8189. https://doi.org/10.1128/JVI.00627-06
McNatt MW, Zang T, Bieniasz PD (2013) Vpu binds directly to Tetherin and displaces it from nascent Virions. PLoS Path 9(4). https://doi.org/10.1371/journal.ppat.1003299
Meyniel-Schicklin L, De Chassey B, André P, Lotteau V (2012) Viruses and interactomes in translation. Mol Cell Proteomics 11(7). https://doi.org/10.1074/mcp.M111.014738
Mi SF, Li Y, Yan JH, Gao GF (2010) Na+/K+-ATPase β1 subunit interacts with M2 proteins of influenza a and B viruses and affects the virus replication. Sci China Life Sci 53(9):1098–1105. https://doi.org/10.1007/s11427-010-4048-7
Mirazimi A, Nilsson M, Svensson L (1998) The molecular chaperone calnexin interacts with the NSP4 enterotoxin of rotavirus in vivo and in vitro. J Virol 72(11):8705–8709
Miura S, Kawana K, Schust DJ, Fujii T, Yokoyama T, Iwasawa Y, Nagamatsu T, Adachi K, Tomio A, Tomio K, Kojima S, Yasugi T, Kozuma S, Taketani Y (2010) CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: a possible mechanism for immune evasion by HPV. J Virol 84(22):11614–11623. https://doi.org/10.1128/JVI.01053-10
Mizushima H, Hijikata M, Asabe SI, Hirota M, Kimura K, Shimotohno K (1994) Two hepatitis C virus glycoprotein E2 products with different C termini. J Virol 68(10):6215–6222
Moll M, Andersson SK, Smed-Sörensen A, Sandberg JK (2010) Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpu interference with CD1d recycling from endosomal compartments. Blood 116(11):1876–1884. https://doi.org/10.1182/blood-2009-09-243667
Montserret R, Saint N, Vanbelle C, Salvay AG, Simorre JP, Ebel C, Sapay N, Renisio JG, Bockmann A, Steinmann E, Pietschmann T, Dubuisson J, Chipot C, Penin F (2010) NMR structure and ion channel activity of the p7 protein from hepatitis C virus. J Biol Chem 285(41):31446–31461
Moradpour D, Penin F (2013) Hepatitis C virus proteins: from structure to function. In: Hepatitis C virus: from molecular virology to antiviral therapy 369, pp 113–142. https://doi.org/10.1007/978-3-642-27340-7_5
Müller M, Prescott EL, Wasson CW, MacDonald A (2015) Human papillomavirus E5 oncoprotein: function and potential target for antiviral therapeutics. Futur Virol 10(1):27–39. https://doi.org/10.2217/fvl.14.99
Münz C (2011) Beclin-1 targeting for viral immune escape. Virus 3(7):1166–1178. https://doi.org/10.3390/v3071166
Nappi VM, Schaefer JA, Petti LM (2002) Molecular examination of the transmembrane requirements of the platelet-derived growth factor β receptor for a productive interaction with the bovine papillomavirus E5 oncoprotein. J Biol Chem 277(49):47149–47159. https://doi.org/10.1074/jbc.M209582200
Neil SJ, Eastman SW, Jouvenet N, Bieniasz PD (2006) HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Path 2(5). https://doi.org/10.1371/journal.ppat.0020039
Neil SJD, Zang T, Bieniasz PD (2008) Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451(7177):425–430. https://doi.org/10.1038/nature06553
Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, Perelson AS (1998) Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282(5386):103–107. https://doi.org/10.1126/science.282.5386.103
Neumann G, Noda T, Kawaoka Y (2009) Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459(7249):931–939
Nieto-Torres JL, DeDiego ML, Álvarez E, Jiménez-Guardeño JM, Regla-Nava JA, Llorente M, Kremer L, Shuo S, Enjuanes L (2011) Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology 415(2):69–82. https://doi.org/10.1016/j.virol.2011.03.029
Nieto-Torres JL, Dediego ML, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Fernandez-Delgado R, Castano-Rodriguez C, Alcaraz A, Torres J, Aguilella VM, Enjuanes L (2014) Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Path 10(5):e1004077. https://doi.org/10.1371/journal.ppat.1004077
Nieto-Torres JL, Verdia-Báguena C, Jimenez-Guardeño JM, Regla-Nava JA, Castaño-Rodriguez C, Fernandez-Delgado R, Torres J, Aguilella VM, Enjuanes L (2015) Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. In press, Virology
Nieva JL, Carrasco L (2015) Viroporins: structures and functions beyond cell membrane permeabilization. Virus 7(10):5169–5171. https://doi.org/10.3390/v7102866
Oelze I, Kartenbeck J, Crusius K, Alonso A (1995) Human papillomavirus type 16 E5 protein affects cell-cell communication in an epithelial cell line. J Virol 69(7):4489–4494
Okada Y, Suzuki T, Sunden Y, Orba Y, Kose S, Imamoto N, Takahashi H, Tanaka S, Hall WW, Nagashima K, Sawa H (2005) Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: role in nuclear egress of viral particles. EMBO Rep 6(5):452–457. https://doi.org/10.1038/sj.embor.7400406
Olmsted RA, Collins PL (1989) The 1A protein of respiratory syncytial virus is an integral membrane protein present as multiple, structurally distinct species. J Virol 63(5):2019–2029
Opella SJ (2015) Relating structure and function of viral membrane-spanning miniproteins. Curr Opin Virol 12:121–125. https://doi.org/10.1016/j.coviro.2015.05.006
Ouyang B, Xie S, Berardi MJ, Zhao X, Dev J, Yu W, Sun B, Chou JJ (2013) Unusual architecture of the p7 channel from hepatitis C virus. Nature 498(7455):521–525
Pan J, Peng X, Gao Y, Li Z, Lu X, Chen Y, Ishaq M, Liu D, DeDiego ML, Enjuanes L, Guo D (2008) Genome-wide analysis of protein-protein interactions and involvement of viral proteins in SARS-CoV replication. PLoS One 3(10). https://doi.org/10.1371/journal.pone.0003299
Parr RD, Storey SM, Mitchell DM, McIntosh AL, Zhou M, Mir KD, Ball JM (2006) The rotavirus enterotoxin NSP4 directly interacts with the caveolar structural protein caveolin-1. J Virol 80(6):2842–2854. https://doi.org/10.1128/JVI.80.6.2842-2854.2006
Pervushin K, Tan E, Parthasarathy K, Lin X, Jiang FL, Yu D, Vararattanavech A, Tuck WS, Ding XL, Torres J (2009) Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Path 5(7)
Petti L, DiMaio D (1992) Stable association between the bovine papillomavirus E5 transforming protein and activated platelet-derived growth factor receptor in transformed mouse cells. Proc Natl Acad Sci U S A 89(15):6736–6740. https://doi.org/10.1073/pnas.89.15.6736
Pham T, Perry JL, Dosey TL, Delcour AH, Hyser JM (2017) The rotavirus NSP4 Viroporin domain is a calcium-conducting Ion Channel. Sci Rep 7. https://doi.org/10.1038/srep43487
Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R (2006) Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proceedings of the Natural Academy of Sciences USA 103(19):7408–7413
Polack FP, Irusta PM, Hoffman SJ, Schiatti MP, Melendi GA, Delgado MF, Laham FR, Thumar B, Hendry RM, Melero JA, Karron RA, Collins PL, Kleeberger SR (2005) The cysteine-rich region of respiratory syncytial virus attachment protein inhibits innate immunity elicited by the virus and endotoxin. Proc Natl Acad Sci U S A 102(25):8996–9001. https://doi.org/10.1073/pnas.0409478102
Popescu CI, Callens N, Trinel D, Roingeard P, Moradpour D, Descamps V, Duverlie G, Penin F, Héliot L, Rouillé Y, Dubuisson J (2011) NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Path 7(2). https://doi.org/10.1371/journal.ppat.1001278
Qi H, Chu V, Wu NC, Chen Z, Truong S, Brar G, Su SY, Du Y, Arumugaswami V, Olson CA, Chen SH, Lin CY, Wu TT, Sun R (2017) Systematic identification of anti-interferon function on hepatitis C virus genome reveals p7 as an immune evasion protein. Proc Natl Acad Sci U S A 114(8):2018–2023. https://doi.org/10.1073/pnas.1614623114
Quistgaard EM, Low C, Moberg P, Guettou F, Maddi K, Nordlund P (2013) Structural and biophysical characterization of the cytoplasmic domains of human BAP29 and BAP31. PLoS One 8(8):e71111. https://doi.org/10.1371/journal.pone.0071111
Ramirez P, Famiglietti M, Sowrirajan B, DePaula-Silva A, Rodesch C, Barker E, Bosque A, Planelles V (2014) Downmodulation of CCR7 by HIV-1 Vpu results in impaired migration and chemotactic Signaling within CD4+ T cells. Cell Rep 7(6):2019–2030. https://doi.org/10.1016/j.celrep.2014.05.015
Ravi LI, Li L, Sutejo R, Chen H, Wong PS, Tan BH, Sugrue RJ (2013) A systems-based approach to analyse the host response in murine lung macrophages challenged with respiratory syncytial virus. BMC Genomics 14:190. https://doi.org/10.1186/1471-2164-14-190
Raynal P, Pollard HB (1994) Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. BBA - Reviews on Biomembranes 1197(1):63–93. https://doi.org/10.1016/0304-4157(94)90019-1
Regan JA, Laimins LA (2008) Bap31 is a novel target of the human papillomavirus E5 protein. J Virol 82(20):10042–10051. https://doi.org/10.1128/JVI.01240-08
Regla-Nava JA, Nieto-Torres JL, Jimenez-Guardeño JM, Fernandez-Delgado R, Fett C, Castaño-Rodríguez C, Perlman S, Enjuanes L, De Diego ML (2015) Severe acute respiratory syndrome coronaviruses with mutations in the E protein are attenuated and promising vaccine candidates. J Virol 89(7):3870–3887. https://doi.org/10.1128/JVI.03566-14
Rixon HW, Brown G, Aitken J, McDonald T, Graham S, Sugrue RJ (2004) The small hydrophobic (SH) protein accumulates within lipid-raft structures of the Golgi complex during respiratory syncytial virus infection. J Gen Virol 85(Pt 5):1153–1165
Rixon HW, Brown G, Murray JT, Sugrue RJ (2005) The respiratory syncytial virus small hydrophobic protein is phosphorylated via a mitogen-activated protein kinase p38-dependent tyrosine kinase activity during virus infection. J Gen Virol 86(Pt 2):375–384
Rodríguez MI, Finbow ME, Alonso A (2000) Binding of human papillomavirus 16 E5 to the 16 kDa subunit c (proteolipid) of the vacuolar H+-ATPase can be dissociated from the E5-mediated epidermal growth factor receptor overactivation. Oncogene 19(33):3727–3732
Rossman JS, Lamb RA (2009) Autophagy, apoptosis, and the influenza virus M2 protein. Cell Host and Microbe 6(4):299–300. https://doi.org/10.1016/j.chom.2009.09.009
Rossman JS, Lamb RA (2011) Influenza virus assembly and budding. Virology 411(2):229–236. https://doi.org/10.1016/j.virol.2010.12.003
Ruch TR, Machamer CE (2011) The hydrophobic domain of infectious bronchitis virus e protein alters the host secretory pathway and is important for release of infectious virus. J Virol 85(2):675–685. https://doi.org/10.1128/JVI.01570-10
Russell RF, McDonald JU, Ivanova M, Zhong Z, Bukreyev A, Tregoning JS (2015) Partial attenuation of respiratory syncytial virus with a deletion of a small hydrophobic gene is associated with elevated interleukin-1beta responses. J Virol 89(17):8974–8981. https://doi.org/10.1128/JVI.01070-15
Safak M, Barrucco R, Darbinyan A, Okada Y, Nagashima K, Khalili K (2001) Interaction of JC virus Agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J Virol 75(3):1476–1486. https://doi.org/10.1128/JVI.75.3.1476-1486.2001
Safak M, Sadowska B, Barrucco R, Khalili K (2002) Functional interaction between JC virus late regulatory agnoprotein and cellular Y-box binding transcription factor, YB-1. J Virol 76(8):3828–3838. https://doi.org/10.1128/JVI.76.8.3828-3838.2002
Sakaguchi T, Tu QA, Pinto LH, Lamb RA (1997) The active oligomeric state of the minimalistic influenza virus M-2 ion channel is a tetramer. Proc Natl Acad Sci U S A 94(10):5000–5005
Sakai A, Claire MS, Faulk K, Govindarajan S, Emerson SU, Purcell RH, Bukh J (2003) The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc Natl Acad Sci U S A 100(20):11646–11651
Saribas AS, Abou-Gharbia M, Childers W, Sariyer IK, White MK, Safak M (2013) Essential roles of Leu/Ile/Phe-rich domain of JC virus agnoprotein in dimer/oligomer formation, protein stability and splicing of viral transcripts. Virology 443(1):161–176. https://doi.org/10.1016/j.virol.2013.05.003
Sariyer IK, Khalili K, Safak M (2008) Dephosphorylation of JC virus agnoprotein by protein phosphatase 2A: inhibition by small t antigen. Virology 375(2):464–479. https://doi.org/10.1016/j.virol.2008.02.020
Sariyer IK, Saribas AS, White MK, Safak M (2011) Infection by agnoprotein-negative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol J 8. https://doi.org/10.1186/1743-422x-8-255
Sastri NP, Viskovska M, Hyser JM, Tanner MR, Horton LB, Sankaran B, Venkataram Prasad BV, Estes MK (2014) Structural plasticity of the coiled-coil domain of rotavirus NSP4. J Virol 88(23):13602–13612. https://doi.org/10.1128/JVI.02227-14
Schapiro F, Sparkowski J, Adduci A, Suprynowicz F, Schlegel R, Grinstein S (2000) Golgi alkalinization by the papillomavirus E5 oncoprotein. J Cell Biol 148(2):305–315. https://doi.org/10.1083/jcb.148.2.305
Schnell JR, Chou JJ (2008) Structure and mechanism of the M2 proton channel of influenza a virus. Nature 451(7178):591–595
Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM (2011) A diverse range of gene products are effectors of the type i interferon antiviral response. Nature 472(7344):481–485. https://doi.org/10.1038/nature09907
Schubert U, Ferrer-Montiel AV, Oblatt-Montal M, Henklein P, Strebel K, Montal M (1996) Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett 398(1):12–18
Seo NS, Zeng CQY, Hyser JM, Utama B, Crawford SE, Kim KJ, Höök M, Estes MK (2008) Integrins α1β1 and α2β1 are receptors for the rotavirus enterotoxin. Proc Natl Acad Sci U S A 105(26):8811–8818. https://doi.org/10.1073/pnas.0803934105
Serrano P, Johnson MA, Almeida MS, Horst R, Herrmann T, Joseph JS, Neuman BW, Subramanian V, Saikatendu KS, Buchmeier MJ, Stevens RC, Kuhn P, Wüthrich K (2007) Nuclear magnetic resonance structure of the N-terminal domain of nonstructural protein 3 from the severe acute respiratory syndrome coronavirus. J Virol 81(21):12049–12060. https://doi.org/10.1128/JVI.00969-07
Shah AH, Sowrirajan B, Davis ZB, Ward JP, Campbell EM, Planelles V, Barker E (2010) Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host and Microbe 8(5):397–409. https://doi.org/10.1016/j.chom.2010.10.008
Shanmugam S, Saravanabalaji D, Yi M (2015) Detergent-resistant membrane association of NS2 and E2 during hepatitis C virus replication. J Virol 89(8):4562–4574. https://doi.org/10.1128/JVI.00123-15
Shapira SD, Gat-Viks I, Shum BOV, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, Hill DE, Regev A, Hacohen N (2009) A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139(7):1255–1267. https://doi.org/10.1016/j.cell.2009.12.018
Sharma K, Surjit M, Satija N, Liu B, Chow VTK, Lai SK (2007) The 3a accessory protein of SARS coronavirus specifically interacts with the 5′UTR of its genomic RNA, using a unique 75 amino acid interaction domain. Biochemistry 46(22):6488–6499. https://doi.org/10.1021/bi062057p
Shen S, Lin PS, Chao YC, Zhang A, Yang X, Lim SG, Hong W, Tan YJ (2005) The severe acute respiratory syndrome coronavirus 3a is a novel structural protein. Biochem Biophys Res Commun 330(1):286–292. https://doi.org/10.1016/j.bbrc.2005.02.153
Simon V, Bloch N, Landau NR (2015) Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol 16(6):546–553. https://doi.org/10.1038/ni.3156
Singh SK, Möckel L, Thiagarajan-Rosenkranz P, Wittlich M, Willbold D, Koenig BW (2012) Mapping the interaction between the cytoplasmic domains of HIV-1 viral protein U and human CD4 with NMR spectroscopy. FEBS J 279(19):3705–3714. https://doi.org/10.1111/j.1742-4658.2012.08732.x
Skasko M, Wang Y, Tian Y, Tokarev A, Munguia J, Ruiz A, Stephens EB, Opella SJ, Guatelli O (2012) HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J Biol Chem 287(1):58–67. https://doi.org/10.1074/jbc.M111.296772
Sou YS, Tanida I, Komatsu M, Ueno T, Kominami E (2006) Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J Biol Chem 281(6):3017–3024. https://doi.org/10.1074/jbc.M505888200
Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T (2007) Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Path 3(7):0962–0971
Strebel K (2014) HIV-1 Vpu - an ion channel in search of a job. Biochim Biophys Acta 1838(4):1074–1081. https://doi.org/10.1016/j.bbamem.2013.06.029
Strebel K, Klimkait T, Martin MA (1988) A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science 241(4870):1221–1223
Su S, Wong G, Shi W, Liu J, Lai ACK, Zhou J, Liu W, Bi Y, Gao GF (2016) Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol 24(6):490–502. https://doi.org/10.1016/j.tim.2016.03.003
Sugden SM, Pham TNQ, Cohen ÉA (2017) HIV-1 Vpu downmodulates ICAM-1 expression, resulting in decreased killing of infected CD4+ T cells by NK cells. J Virol 91(8). https://doi.org/10.1128/JVI.02442-16
Sun L, Hemgård GV, Susanto SA, Wirth M (2010) Caveolin-1 influences human influenza A virus (H1N1) multiplication in cell culture. Virol J 7. https://doi.org/10.1186/1743-422X-7-108
Surti T, Klein O, Aschheim K, DiMaio D, Smith SO (1998) Structural models of the bovine papillomavirus E5 protein. Proteins Struct Funct Genet 33(4):601–612. https://doi.org/10.1002/(SICI)1097-0134(19981201)33:4<601::AID-PROT12>3.0.CO;2-I
Suzuki T, Okada Y, Sembat S, Orba Y, Yamanouchii S, Endo S, Tanaka S, Fujita T, Kuroda S, Nagashima K, Sawa H (2005) Identification of FEZ1 as a protein that interacts with JC virus agnoprotein and microtubules: role of agnoprotein-induced dissociation of FEZ1 from microtubules in viral propagation. J Biol Chem 280(26):24948–24956. https://doi.org/10.1074/jbc.M411499200
Suzuki T, Orba Y, Makino Y, Okada Y, Sunden Y, Hasegawa H, Hall WW, Sawa H (2013) Viroporin activity of the JC polyomavirus is regulated by interactions with the adaptor protein complex 3. Proc Natl Acad Sci U S A 110(46):18668–18673. https://doi.org/10.1073/pnas.1311457110
Suzuki T, Orba Y, Malcino Y, Okada Y, Sunden Y, Kimura T, Hasegawa H, Sata T, Hall WW, Sawa H (2010a) Disruption of intracellular vesicular trafficking by Agnoprotein is essential for Viroporin activity and JC virus replication. J Neurovirol 16:84–84
Suzuki T, Orba Y, Okada Y, Sunden Y, Kimura T, Tanaka S, Nagashima K, Hall WW, Sawa H (2010b) The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Path 6(3):e1000801. https://doi.org/10.1371/journal.ppat.1000801
Suzuki T, Semba S, Sunden Y, Orba Y, Kobayashi S, Nagashima K, Kimura T, Hasegawa H, Sawa H (2012) Role of JC virus agnoprotein in virion formation. Microbiol Immunol 56(9):639–646. https://doi.org/10.1111/j.1348-0421.2012.00484.x
Takeuchi K, Lamb RA (1994) Influenza virus M2 protein ion channel activity stabilizes the native form of fowl plague virus hemagglutinin during intracellular transport. J Virol 68(2):911–919
Tan YJ, Lim SG, Hong W (2006) Understanding the accessory viral proteins unique to the severe acute respiratory syndrome (SARS) coronavirus. Antivir Res 72(2):78–88. https://doi.org/10.1016/j.antiviral.2006.05.010
Tan YJ, Teng E, Shen S, Tan THP, Goh PY, Fielding BC, Ooi EE, Tan HC, Lim SG, Hong W (2004) A novel severe acute respiratory syndrome coronavirus protein, U274, is transported to the cell surface and undergoes endocytosis. J Virol 78(13):6723–6734. https://doi.org/10.1128/JVI.78.13.6723-6734.2004
Tan YJ, Tham PY, Chan DZL, Chou CF, Shen S, Fielding BC, Tan THP, Lim SG, Hong W (2005) The severe acute respiratory syndrome coronavirus 3a protein up-regulates expression of fibrinogen in lung epithelial cells. J Virol 79(15):10083–10087. https://doi.org/10.1128/JVI.79.15.10083-10087.2005
Tapia LI, Shaw CA, Aideyan LO, Jewell AM, Dawson BC, Haq TR, Piedra PA (2014) Gene sequence variability of the three surface proteins of human respiratory syncytial virus (HRSV) in Texas. PLoS One 9(3). https://doi.org/10.1371/journal.pone.0090786
Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick SW (2008) An in vivo map of the yeast protein interactome. Science 320(5882):1465–1470. https://doi.org/10.1126/science.1153878
Tate JE, Burton AH, Boschi-Pinto C, Parashar UD, Agocs M, Serhan F, De Oliveira L, Mwenda JM, Mihigo R, Ranjan Wijesinghe P, Abeysinghe N, Fox K, Paladin F (2016) Global, regional, and National Estimates of rotavirus mortality in children <5 years of age, 2000-2013. Clin Infect Dis 62:S96–S105. https://doi.org/10.1093/cid/civ1013
Taylor G, Wyld S, Valarcher JF, Guzman E, Thom M, Widdison S, Buchholz UJ (2014) Recombinant bovine respiratory syncytial virus with deletion of the SH gene induces increased apoptosis and pro-inflammatory cytokines in vitro, and is attenuated and induces protective immunity in calves. J Gen Virol 95:1244–1254. https://doi.org/10.1099/Vir.0.064931-0
Taylor JA, O'Brien JA, Yeager M (1996) The cytoplasmic tail of NSP4, the endoplasmic reticulum-localized non-structural glycoprotein of rotavirus, contains distinct virus binding and coiled coil domains. EMBO J 15(17):4469–4476
Teoh KT, Siu YL, Chan WL, Schluter MA, Liu CJ, Peiris JS, Bruzzone R, Margolis B, Nal B (2010) The SARS coronavirus E protein interacts with PALS1 and alters tight junction formation and epithelial morphogenesis. Mol Biol Cell 21(22):3838–3852. https://doi.org/10.1091/mbc.E10-04-0338
Terwilliger EF, Cohen EA, Lu Y, Sodroski JG, Haseltine WA (1989) Functional role of human immunodeficiency virus type 1 vpu. Proc Natl Acad Sci U S A 86(13):5163–5167
To J, Surya W, Fung TS, Li Y, Verdià-Bàguena C, Queralt-Martin M, Aguilella VM, Liu DX, Torres J (2017) Channel-inactivating mutations and their revertant mutants in the envelope protein of infectious bronchitis virus. J Virol 91(5). https://doi.org/10.1128/JVI.02158-16
To J, Surya W, Torres J (2016) Targeting the channel activity of Viroporins. Adv Protein Chem Struct Biol 104:307–355. https://doi.org/10.1016/bs.apcsb.2015.12.003
Tomakidi P, Cheng H, Kohl A, Komposch G, Alonso A (2000) Connexin 43 expression is downregulated in raft cultures of human keratinocytes expressing the human papillomavirus type 16 E5 protein. Cell Tissue Res 301(2):323–327
Torres J, Maheswari U, Parthasarathy K, Ng LF, Liu DX, Gong XD (2007) Conductance and amantadine binding of a pore formed by a lysine-flanked transmembrane domain of SARS coronavirus envelope protein. Protein Sci 16(9):2065–2071. https://doi.org/10.1110/ps.062730007
Torres J, Parthasarathy K, Lin X, Saravanan R, Kukol A, Ding XL (2006) Model of a putative pore: the pentameric α-helical bundle of SARS coronavirus E protein in lipid bilayers. Biophys J 91(3):938–947
Triantafilou K, Kar S, Vakakis E, Kotecha S, Triantafilou M (2013) Human respiratory syncytial virus viroporin SH: a viral recognition pathway used by the host to signal inflammasome activation. Immunology 140:87–88
Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J (2008) The interferon-induced protein BST-2 restricts HIV-1 release and is Downregulated from the cell surface by the viral Vpu protein. Cell Host and Microbe 3(4):245–252. https://doi.org/10.1016/j.chom.2008.03.001
Varga ZT, Grant A, Manicassamy B, Palese P (2012) Influenza virus protein pb1-f2 inhibits the induction of type I interferon by binding to mavs and decreasing mitochondrial membrane potential. J Virol 86(16):8359–8366. https://doi.org/10.1128/JVI.01122-12
Varga ZT, Palese P (2011) The influenza a virus protein PB1-F2: killing two birds with one stone? Virulence 2(6):542–546
Vassena L, Giuliani E, Koppensteiner H, Bolduan S, Schindler M, Doria M (2015) HIV-1 Nef and Vpu interfere with L-selectin (CD62L) cell surface expression to inhibit adhesion and signaling in infected CD4<sup>+</sup> T lymphocytes. J Virol 89(10):5687–5700. https://doi.org/10.1128/JVI.00611-15
Venkatesh S, Bieniasz PD (2013) Mechanism of HIV-1 Virion entrapment by Tetherin. PLoS Path 9(7). https://doi.org/10.1371/journal.ppat.1003483
Verdia-Baguena C, Nieto-Torres JL, Alcaraz A, DeDiego ML, Torres J, Aguilella VM, Enjuanes L (2012) Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology 432(2):485–494. https://doi.org/10.1016/j.virol.2012.07.005
Vieyres G, Dubuisson J, Pietschmann T (2014) Incorporation of hepatitis C virus E1 and E2 glycoproteins: the keystones on a peculiar virion. Virus 6(3):1149–1187. https://doi.org/10.3390/v6031149
Vigan R, Neil SJD (2010) Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J Virol 84(24):12958–12970. https://doi.org/10.1128/JVI.01699-10
von Brunn A, Teepe C, Simpson JC, Pepperkok R, Friedel CC, Zimmer R, Roberts R, Baric R, Haas J (2007) Analysis of intraviral protein-protein interactions of the SARS coronavirus ORFeome. PLoS One 2(5). https://doi.org/10.1371/journal.pone.0000459
Wang CK, Pan L, Chen J, Zhang M (2010) Extensions of PDZ domains as important structural and functional elements. Protein and Cell 1(8):737–751. https://doi.org/10.1007/s13238-010-0099-6
Wang L, Fu B, Li W, Patil G, Liu L, Dorf ME, Li S (2017) Comparative influenza protein interactomes identify the role of plakophilin 2 in virus restriction. Nat Commun 8. https://doi.org/10.1038/ncomms13876
Watanabe R, Leser GP, Lamb RA (2011) Influenza virus is not restricted by tetherin whereas influenza VLP production is restricted by tetherin. Virology 417(1):50–56. https://doi.org/10.1016/j.virol.2011.05.006
Weiner LB, Polak MJ, Masaquel A, Mahadevia PJ (2011) Cost-effectiveness of respiratory syncytial virus prophylaxis with Palivizumab among preterm infants covered by Medicaid in the United States. Value Health 14(3):A118–A118
Wetherill LF, Holmes KK, Verow M, Müller M, Howell G, Harris M, Fishwick C, Stonehouse N, Foster R, Blair GE, Griffin S, Macdonald A (2012) High-risk human papillomavirus E5 oncoprotein displays channel-forming activity sensitive to small-molecule inhibitors. J Virol 86(9):5341–5351. https://doi.org/10.1128/JVI.06243-11
Willey RL, Buckler-White A, Strebel K (1994) Sequences present in the cytoplasmic domain of CD4 are necessary and sufficient to confer sensitivity to the human immunodeficiency virus type 1 Vpu protein. J Virol 68(2):1207–1212
Willey RL, Maldarelli F, Martin MA, Strebel K (1992) Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J Virol 66(12):7193–7200
Wong SLA, Chen Y, Chak MC, Chan CSM, Chan PKS, Chui YL, Kwok PF, Waye MMY, Tsui SKW, Chan HYE (2005) In vivo functional characterization of the SARS-coronavirus 3a protein in drosophila. Biochem Biophys Res Commun 337(2):720–729. https://doi.org/10.1016/j.bbrc.2005.09.098
Wozniak AL, Griffin S, Rowlands D, Harris M, Yi M, Lemon SM, Weinman SA (2010) Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog 6(9):e1001087. https://doi.org/10.1371/journal.ppat.1001087
Xu A, Bellamy AR, Taylor JA (2000) Immobilization of the early secretory pathway by a virus glycoprotein that binds to microtubules. EMBO J 19(23):6465–6474
Ye F, Zhang M (2013) Structures and target recognition modes of PDZ domains: recurring themes and emerging pictures. Biochem J 455(1):1–14. https://doi.org/10.1042/BJ20130783
Yi M, Ma Y, Yates J, Lemon SM (2007) Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chirneric hepatitis C virus. J Virol 81(2):629–638
Yuan X, Yao Z, Wu J, Zhou Y, Shan Y, Dong B, Zhao Z, Hua P, Chen J, Cong Y (2007) G1 phase cell cycle arrest induced by SARS-CoV 3a protein via the Cyclin D3/pRb pathway. Am J Respir Cell Mol Biol 37(1):9–19. https://doi.org/10.1165/rcmb.2005-0345RC
Zebedee SL, Lamb RA (1989) Growth restriction of influenza a virus by M<inf>2</inf> protein antibody is genetically linked to the M<inf>1</inf> protein. Proc Natl Acad Sci U S A 86(3):1061–1065
Zhang R, Wang K, Lv W, Yu W, Xie S, Xu K, Schwarz W, Xiong S, Sun B (2014) The ORF4a protein of human coronavirus 229E functions as a viroporin that regulates viral production. Biochim Biophys Acta Biomembr 1838(4):1088–1095. https://doi.org/10.1016/j.bbamem.2013.07.025
Zhang R, Wang K, Ping X, Yu W, Qian Z, Xiong S, Sun B (2015) The ns12.9 accessory protein of human coronavirus OC43 is a viroporin involved in virion morphogenesis and pathogenesis. J Virol 89(22):11383–11395. https://doi.org/10.1128/JVI.01986-15
Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, Mitchell T, Miller P, Dean RA, Gerstein M, Snyder M (2001) Global analysis of protein activities using proteome chips. Science 293(5537):2101–2105. https://doi.org/10.1126/science.1062191
Zhu P, Liang L, Shao X, Luo W, Jiang S, Zhao Q, Sun N, Zhao Y, Li J, Wang J, Zhou Y, Zhang J, Wang G, Jiang L, Chen H, Li C (2017) Host cellular protein TRAPPC6AΔ interacts with influenza a virus M2 protein and regulates viral propagation by modulating M2 trafficking. J Virol 91(1). https://doi.org/10.1128/JVI.01757-16
Zou P, Wu F, Lu L, Huang JH, Chen YH (2009) The cytoplasmic domain of influenza M2 protein interacts with caveolin-1. Arch Biochem Biophys 486(2):150–154. https://doi.org/10.1016/j.abb.2009.02.001
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
To, J., Torres, J. (2018). Beyond Channel Activity: Protein-Protein Interactions Involving Viroporins. In: Harris, J., Bhella, D. (eds) Virus Protein and Nucleoprotein Complexes. Subcellular Biochemistry, vol 88. Springer, Singapore. https://doi.org/10.1007/978-981-10-8456-0_15
Download citation
DOI: https://doi.org/10.1007/978-981-10-8456-0_15
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8455-3
Online ISBN: 978-981-10-8456-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)