
Abstract
Radiotherapy is a commonly used treatment for cancer and is usually given in varying doses. Mathematical modelling of radiation effects traditionally means the modelling or estimation of cell-kill due to its direct exposure to irradiation and sometimes ignoring other multiple direct/indirect effects. However, advances in molecular biology have expanded this classical view and it is now realized that in addition to cell-death, signals produced by irradiated cells can further influence the behavior of non-irradiated cells or organisms in several ways. Consequently, it has now wider implications in multiple areas making it relevant for further exploration, both experimentally and mathematically. Here, we provide a brief overview of a hybrid multiscale mathematical model to study the direct and indirect effects of radiation and its implications in clinical radiotherapy, experimental settings and radiation protection.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


1 Introduction
Radiotherapy (RT) is currently part of the treatments of 40% of cancer patients in the UK. This comprises patients receiving external beam RT , particle irradiation, molecular radiotherapy and brachytherapy. Independent of the treatment modality used, a treatment plan or dose simulation is performed for each patient based on computed tomography or standard x-ray images to conform the radiation dose delivered to the tumour and to spare the surrounding normal tissues and organs at risk. Although through this process the physical dose delivered may be calculated with high precision using state of the art dose calculations, the physical dose will always remain a surrogate for the biological effects induced. Understanding and quantifying these biological effects is, however, critical for prediction and evaluation of treatment response. Quantification may range spatially from sub-cellular level (DNA damage), to the cellular scale in terms of radiation induced cell kill, to tissue and organ level (organ functionality), the overall patient response (survival), and finally the impact of radiation on a treated population as a whole. This implies that also the temporal scale on which these effects are observed and impact may vary from minutes to years. Computational modelling may help to better understand, quantify and predict the biological response on any of these spatio-temporal scales and may therefore bridge the gap between biological effects observed in vitro and in vivo. In contrast to analytical dose-response models, systems biology simulations model the biological response in a time and space dependent manner allowing for a detailed description of the dynamic behaviors. As such, systems biology represents a holistic approach that describes complex biological systems using a number of interdependent components that result in the function and behavior of the system. Depending on the spatio-temporal scales of interest, the interaction of genes, proteins, cells or larger sub-structures within a tissue or organism may be described. For example, in the case of radiation response modelling for cancer therapy applications models may start from describing individual cells with intra- and intercellular processes being governed by a set of predefined rules or a system of ODEs (ordinary differential equations). By simulating a large number of these interacting cells, insight into emergent tissue level phenomena can be achieved.
2 Multiscale Modelling to Study Radiation Effects
2.1 Mathematical Model: Multiscale Approach
A number of modelling approaches exists within the framework of systems biology simulations, but in general two groups can be defined: discrete and continuum-based approaches. Continuum-based models describe the system entities by a set of (partial) differential equations. Cells are not modelled directly but are described in terms of cell densities that fluctuate with time. Multiple cell types may be described by interconnected sets of equations. Distributions of nutrients, oxygen, or signaling molecules are modelled by iteratively solving the respective reaction-diffusion equation.
The most basic approach of discrete models is a cellular automaton (CA). CA models represent cells as discrete voxels/pixels on a fixed grid and cellular processes are driven by a set of pre-defined rules and cell properties. As such, CA requires defining a regular lattice of a fixed size, on which initially some pixel/voxel are occupied by ‘cells’ (initialization step). Each cell can be in one of several predefined states, these could be different cell cycle stages, or simply ‘alive’ vs. ‘dead’. The stages of all cells are then updated in each time step according to the rules defined. For example, cell cycle progression can be controlled by assigning an individual clock to each cell that is incremented in each simulation time step until a threshold time is reached upon which the cell divides into two daughter cells. In general, all rules are pre-defined in CA, meaning that they remain unchanged throughout the simulation . Given their simple implementation, CA models have widely been used to simulate a number of different scenarios, such as tumor progression (Anderson & Chaplain, 1998; Kansal et al., 2000; Patel et al., 2001; Turner & Sherratt, 2002), or cellular treatment response modelling in vitro (Ribba et al., 2006; Richard et al., 2007). A comprehensive review on the use of CA for cancer modelling applications has been provided by Moreira and Deutsch (2002).
An alternative individual based approach is using a multiscale cellular Potts model (CPM) or the Glazier-Graner-Hogeweg (GGH ) approach (Glazier et al., 2007). The GGH model is very well-suited to model individual objects (such as cancer cells) and interactions with the cellular properties that evolve with respect to time and space (Andasari et al., 2012). Each cell is considered as a collection of lattice pixels with a common index marker and is represented as a spatially extended domain on a fixed lattice, either a 3D Cartesian lattice or a 3D hexagonal lattice. The cell’s behaviors and interactions are defined through the local minimization of effective energies depending on cell and pixel configurations. By minimizing energy via the Modified Metropolis Algorithm, CPM recovers a linear relation between force and a cell’s velocity and hence allows for the translation of lab-measurable cellular characteristics into model parameters. Regardless of choice of computational approach, each method should give qualitatively similar results for the same biologically-determined classes of objects, behaviors and interactions. Any observed discrepancies between methods can used to veto and/or improve modelling methods (Powathil et al., 2015).
The advantage of CA and CPM approaches is their detailed description and their capability to describe the progression of individual cells and the population as a whole. Depending on the detail of cellular properties accounted for, the number of cells and/or the simulated time span may, however, be limited. To limit computational cost there has to be a trade-off between the level of detail and the overall number of cells per simulation . Hybrid models between CA or CPM and continuum models have been developed to overcome some of the limitations of these models. In hybrid models the entire multiscale is simulated over a finite period of time and the simulation of cells is controlled in a CA fashion as individual grid points. In addition to this, micro environmental dynamics such as oxygen or nutrient diffusion are modelled using a continuum-based approach. At every time step, a finite difference scheme is evaluated and the corresponding nutrient concentration levels are updated and assigned at each grid position. Timescale increments may, however, be quite different for these two model combinations (hours for cell division vs. sub-second increments for finite differences) requiring careful synchronization upon implementation. Recently, Powathil et al. developed a hybrid multiscale cellular automaton approach to model cancer progression and used the model to study the effects of cell-cycle dependent chemotherapeutic drugs alone and in combination with radiation therapy (Powathil et al., 2012a, 2013). Examples of hybrid CA and CPM are shown in Fig. 5.1.

Figure adapted from (Powathil et al., 2015). Spatial cell and vessel distributions of simulations using a hybrid multiscale CA (i) or a CPM (ii) are shown. In both model, colours represent various stages of cell-cycle and the simulation is started from a single cell at 0 h. These are for the (i): G1 (blue), S-G2 -M (green), resting (magenta), hypoxic cells in G1 (rose), hypoxic cells in S-G2 -M (yellow) and hypoxic cells in resting (silver). (ii) Plots from hybrid CPM (using Compucell3D) and the colour legend shows the types of the tumour cells; For (ii) the color legend indicates the cell type: 1- surrounding medium, 2- G2 phase, 3- G1 phase, 4- vessel cross sections, 5- hypoxic G2 phase, 6- hypoxic G1 phase and 7- resting cells
2.2 Multiscale Model Implementation
As mentioned previously, there is always a tradeoff to be made between the number of cells and detail of the inter- and intracellular processes modelled. Depending on the problem to be simulated, the choice of model is the essential first step towards an efficient implementation. A direct implementation of a CA model is ideally optimized for computational performance to maximize its quality and significance. The easiest way to implement a CA is by defining a fixed sized array of designated cell positions. The respective cellular or micro environmental properties (occupancy, cell cycle stage, oxygen level, etc.) are directly assigned to each element of this array. Each time frame loops over all array elements and potentially updates relevant properties. Although this describes a very simple implementation, it means that a fixed number of positions have to be processed regardless of their occupancy by cells. Obviously, this represents a very poor implementation in terms of computational efficacy with the limiting factor of the simulation being the overall size of the array. One option to speed up this calculation has been proposed by Poleszczuk and Enderling (2014) who continuously increase the computational domain upon colony growth. However, this may come at the cost of having to reorganize data structures in memory throughout the simulation . A more scalable method which aims for large scale problems on high performance computing hardware was proposed by Brüningk et al. (2018). In this work, the spatial cell grid and the physical properties of the cells are stored separately in memory in a list-like structure which grows along the cells at constant costs. The logic separation of the data allows for a quicker pass-through of the cell arrangement and facilitates a parallel or distributed implementation of the CA .
The computational simulations of the multiscale CPM are implemented in the CompuCell3D framework developed by Glazier et al. (see http://www.compucell3d.org for full details) (Powathil et al., 2015, 2016). Here, cells are represented by a collection of pixels in a 2D lattice and as in hybrid CA model, the cellular proliferation is governed by the internal cell-cycle dynamics modelled using the kinetic equations as given in Fig. 5.2. These set of equations are solved in each Monte Carlo time step using SBML solvers such as Bionetsolver (Powathil et al., 2015). The evolution of concentration fields such as oxygen concentration is incorporated into the CompuCell3D as a diffusive chemical field that follows the respective partial differential equation as given in Fig. 5.2. Further details of both the simulation approaches that are used to study the multiscale mathematical model and the relevant parameter values can be found in recent papers by Powathil et al. (2012a, 2013, 2014).

Figure showing various processes involved in the simulation. Top: Plot of the concentration profiles of the various intracellular proteins and the cell-mass over a period of 200 h for one automaton cell in the model. This is obtained by solving the system of equations shown under ‘Cell-cycle dynamics’, with the relevant parameter values from Powathil et al. (2012a). Middle: Representative realization of the spatial distribution of oxygen (K(x, t)) obtained by solving the reaction-diffusion equation . This equation describes the dynamic change of oxygenation due to oxygen diffusion (first term), oxygen production at vessel locations (second term), and oxygen consumption by the cells (third term). Bottom: Radiation effects are modeled by calculating the surviving fraction S as a function of dose delivered, cell cycle stage (γ), and oxygenation level (OMF). Here the oxygen modification factor (OMF) is calculated as the ratio of the oxygen enhancement rations (OER) at the position of the cell (OER(pO2)), and the maximum OER, OERM. Here, pO2 (x) is the oxygen concentration at position x, and Km is the oxygen level at which the OER is at half its maximum level
2.3 Applications of Systems Biology Simulations
In the previous sections a number of modelling approaches have been introduced. Each of these models has its specific advantages and shortcomings, and which modelling approach is suited for the purpose is highly dependent on the question to be answered. Multiscale models can provide a better understanding of the underlying biological reaction chain and its influence on the emerging system response. As such, these simulations allow studying and weighting the importance of different contributing mechanisms and pathways to the overall response using a sensitivity analysis. A (variance-based) sensitivity analysis (Satelli et al., 2010) may point out which model parameters are most influential and thus crucial for defining the uncertainty of the simulation result. This is important since the analysis may flag up parameters that are essential to be validated experimentally, whereas rough estimates may be sufficient in other cases saving time and resources.
Moreover, systems approaches, allow analyzing the influence of a heterogeneous subset of different cell types and to study their interaction. Although it may be possible to experimentally validate single cell type based simulations, experiments involving multiple cell types are difficult and simulations may help to better predict more realistic scenarios of heterogeneous populations. Within such more realistic scenarios, it is then possible to identify and optimize radiation schedules for cancer treatments, potentially in combination with other treatment modalities such as targeted drugs or chemotherapy . In particular, it may be possible to include patient specific information allowing for a personalized treatment optimization. In the following we will give two examples of multiscale models and their application.
3 Modelling Cellular Response to Radiotherapy: Simulation and Validation
In this section, we describe the components of a simulation framework that models radiation response at a cellular level in the range of therapeutic doses as used clinically for radiotherapy treatments (1–5 Gy, 1–4 Gy/min). Such a framework consists of four main parts: A simulation of the cellular microenvironment, an implementation of normal cell growth within this environment, treatment delivery, and response modelling. Different modelling approaches are chosen for each of these components to minimize computation time (hybrid CA model).
3.1 Setting the Scene: Modelling Cellular Growth
Normal cell growth is simulated using the CA approach. Cells are modelled individually as elements of a two or three dimensional lattice allowing for modelling of simple in vitro experiments or a small part of a tumor in vivo. Lattice dimensions are defined by the geometry and type of cell simulated. Here these have been adapted to match the growth response of HCT116 cells, a commonly used human colorectal cancer cell line. We use a pixel size of 12 × 12μm on a 3000 × 3000 big lattice to model cells in two dimensional 6-well plates (well diameter 34.8 mm). For three dimensional models, a smaller grid of 200 × 200 × 200 voxel of the same size is used.
To enable the proliferation of the cells in the CA model we need to define specific rules: Here, each cell follows a four phase cell cycle (G1, S, G2, M phases). Depending on the level of detail to be simulated, cell cycle progression may be controlled by a set of ODEs modelling the temporal evolution of a number of cell cycle protein complexes as shown in Fig. 5.3. This approach, which was first proposed by Tyson and Novak (2001; Novak & Tyson, 2003), has the advantage of closely modelling the biological reaction chain underlying the cell cycle, where different cycle stages are controlled by the level of cyclin complexes. These cyclin complex levels may be influenced by external factors such as the oxygenation level of the cell (here modelled by assuming the activation of the HIF pathway).

Probability driven response cascade used to model the dynamic response of cells to radiation mimicking the processes of mitotic catastrophe. Upon irradiation, a random number N is drawn for each cell and compared to the respective surviving fraction S calculated according to Eq. (5.2). If the cell is drawn to die (N > S), it is assigned a time to cell death and continues to proliferate until for this time. Once the cell reaches M-phase, it exits the cell cycle at probability pdivison. These cells then either become senescent (at probability psenescent) or turn into a giant cell that increases in mass but does not divide. Once the delay to cell death is up, the cell and potential daughter cells are removed from the computational domain
If detailed pathway dependence is not needed, a much simpler and computationally effective method for cell cycle implementation is a cell cycle timer that is assigned to each cell. This means that the cell cycle is characterized by cycle phase specific transition times to the following stage. Once the predefined doubling time is reached the cell divides. These parameters can either be measured experimentally, or estimated numbers can be used if validation data is not available. Typically, cell doubling times range around 24 h for tumor cells, but may be significantly longer for normal cells depending on the specific cell type. The computation time-step after which the cell cycle timers are incremented may therefore be in the order of several minutes to hours. Here we used a doubling time of 19.5 h, and assumed an infinite number of possible divisions for the cells.
Independent of the cell cycle model used cells progress through their cycle and will divide into two identical daughter cells at the end of M-phase. The position of the new cell is randomly selected from the neighborhood of its parental cell. The type of neighborhood considered is alternated between von Neumann and Moore neighborhoods to avoid generating cell distribution patterns matching the specific neighborhood (Powathil et al., 2012a, 2013). Cell colonies will thus grow in circular patterns and no further cell migration is assumed to take place. It is defined that, if there are no free neighboring lattice positions available, the cell will enter the reversible quiescent stage G0. Cells in G0 arrest their cell cycle and suspend their cellular activities until neighboring spaces are vacated. Once entering G0, a timer is started for the cell indicating the G0 duration. If the timer exceeds a critical limit, the cell is labeled as necrotic meaning that its position is not vacated but the cell is considered ‘dead’.
3.2 Modelling the Cellular Microenvironment
The cellular microenvironment controls the oxygen and nutrient supply of the cells and therefore influences the cells’ treatment and growth response. In a simple model, oxygen may either be supplied by a number of blood vessels scattered over the computational grid (Powathil et al., 2013), or modelled as being supplied externally from the surrounding medium, i.e. from outside a 3D cell aggregate or uniformly in the case of 2D cultures in a dish. The oxygen distribution K(x,t) may then be calculated by iteratively solving the reaction-diffusion equation given in Fig. 5.2 using finite difference method. Here, the change in oxygen level at each position is calculated from contributions of oxygen diffusion, oxygen production from potential vessel locations, and oxygen consumption by the cells. The parameters used are the diffusion coefficient of oxygen in the respective tissue DK, the oxygen production rate r(x) at vessel locations of cross section m(x), and the oxygen consumption rate of the cells, Φ, if a cell is present at position x (cells(x,t)). To iteratively solve the diffusion equation it is essential to use meaningful boundary conditions. These may be full oxygenation outside the cell aggregate (no vessels), or no-flux boundary conditions are imposed in the case of vessel distributions if an internal part of a tumor is simulated. An example of an oxygen distribution originating from a number of randomly distributed blood vessels is shown in Fig. 5.2. Gradients of other diffusive molecules, such as nutrients, drugs, or signaling molecule s can be modelled accordingly.
3.3 Treatment Delivery and Response Modelling
Since the cell cycle timer increments are on the order of minutes to hours, irradiation using therapeutic dose rates of several Gray per minute may be simulated by assigning the total dose delivered to each cell within one time increment. For modelling cell survival of each individual cell, the probability of cell survival, S, at the specific dose level, d, is first calculated using the linear quadratic (LQ) cell survival model (Fowler, 1989):
The LQ-model uses two cell line and treatment modality depended parameters, α and β. These parameters were referred to contributions of cell kill originating from double and single strand breaks in the original publication of the LQ model, but their interpretation has since then been debated. In a systems biology simulation , each cell is assigned specific values for α and β according to the intrinsic radio sensitivity of the cell type modelled. If a large number of cells from the same type are simulated, values displaying small differences from the known means may be assigned to each cell while maintaining the overall average for the population as a whole.
In addition to the intrinsic radio sensitivity of the cell type, the cells cycle stage also influences radio sensitivity due to differences in pre-activated DNA repair pathways (Lauber et al., 2012). One option to account for this is the inclusion of a cell cycle stage specific weighting factor γ in (5.1) (Powathil et al., 2012a).
Microenvironmental factors, such as the oxygenation level, may further influence the radio sensitivity of the cells. This may be accounted for by multiplying the radiation dose with an oxygen level dependent weighting factor. This oxygen modification factor (OMF) ranges between 1 and 3 depending on the current partial oxygen pressure at the cells location. The respective equation is shown in the summary Fig. 5.2. The OMF introduces a higher probability of cell survival in the absence of oxygen and increases the cell-kill if the cell is well oxygenated (Powathil et al., 2012b).
The LQ-model generally describes clonogenic cell survival (Franken et al., 2006), meaning that it gives an estimation of the overall proportion of a treated cell population that are still able to successfully undergo division and may therefore be the origin of increasing tumor burden. The LQ-model does not inform the dynamics of the cell kill, or how quickly cells which are unable to form a colony will be unable to divide and/or die. To account for this problem, we are using a probability driven response cascade to model the dynamics of radiation induced mitotic catastrophe which is the major pathway involved in radiation-induced cell kill (Lauber et al., 2012) (see Fig. 5.3). In this cascade, a random number N between 0 and 1 is drawn for each cell in the first time step post irradiation. Whereas cell with N ≤ S continue to cycle normally after a short delay to allow damage repair, cells with a number drawn that exceeds the survival threshold are labelled as non clonogenic. Non clonogenic cells are assigned a delay time to cell death randomly sampled from an exponential distribution. These cells will continue to progress though their cell cycle, but in M-phase will not divide at a probability pdivision, but become either senescent (not proliferating), or giant cell (increasing in size, but no division). Once the delay to cell death has been reached, the cell is removed from the computational grid if connected to empty space, or labeled as necrotic if at the center of a colony. It is important to note, that the threshold probabilities and delay times used , may be cell line and dose range dependent. This makes it essential to validate the simulation framework experimentally before making predictions.
3.4 Simulation Validation
Experimental validation is an essential part of any model to allow making meaningful predictions and to minimize the number of free parameters . In the case of systems biology simulations, validation may happen on any of the spatio-temporal scales used, resulting in a potentially very large number of datasets needed to fully validate every part of the model. Whereas simulations modelling in vitro scenarios may be relatively easy to validate, it is much more difficult in more complex simulations of realistic, in vivo-like cases. In vitro experiments used to calibrate a simulation to model the behavior of a particular cell type include cell size measurements, cell cycle analysis using Flow cytometry, cell survival evaluation with clonogenic and/or cell viability assays, as well as cellular growth curves of treated and untreated populations. 3D cell aggregates such as spheroids may be used to easily validate cellular behavior in 3D geometries and to include micro environmental aspects in the validation data. Histological analysis of spheroid sections may be used to validate oxygen distributions, as well as proportions of proliferating, cell-cycle arresting, and dead cells. Finally, tissue specimens from excised tumors can be stained to quantify vessel and oxygen distributions, proportions of tumor and normal cells and their viability.
Recently, Brüningk et al. (2018) presented a validated hybrid CA model for studying the effects of combinations of radiation and hyperthermia (heat) treatments. In this study, the response of a commonly used human colorectal cancer cell line (HCT116) was analyzed using clonogenic assays, and manually counted growth curves. For manual growth curve counts, samples were cultured in 6-well plates resulting in several hundreds of thousands to millions of cells per well. This means that even if experimental reference data is available, a direct model calibration is only possible if a large number of cells can be simulated which may be difficult in a CA model. In their model, Brüningk et al. first calibrated their framework to normal cellular growth by adapting cell size, doubling time, and cell cycle distribution. Then, growth curves of irradiated samples were used to adapt parameters in the response cascade used to model the radiation response dynamics. Finally, the simulation was successfully validated on another set of cellular growth curves for single and multiple treatment fractions without making further changes to the underlying model.
4 Modelling Indirect Effects: Radiation-Induced Bystander Effects
Recent advances in radiobiology expanded beyond studying the direct (targeted) effects of radiation and explored various factors associated with non- targeted effects of radiation, including the phenomena known as the “bystander effects”, where the signals produced by irradiated cells influence the behavior of non- irradiated cells (Blyth & Sykes, 2011; Prise & O’Sullivan, 2009; Mothersill & Seymour, 2004; Morgan, 2003a, b). At low radiation doses, since the direct cell kill is relatively low, the non-targeted effects are shown to play a major role in cellular response to radiation (Mothersill & Seymour, 2004; Munro, 2009; Prise & O’Sullivan, 2009). This has major implications in several areas, especially, in clinical radiotherapy and radioecology, where cells or organisms are exposed to varying radiation levels, especially at low dose rates.
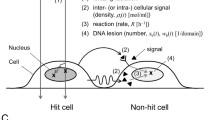
Although it is difficult to separate direct effects from bystander effects, there are several experimental studies that investigate these effects in various ways (Azzam et al., 2000; Fernandez-Palomo et al., 2015; Hu et al., 2006; Lorimore et al., 2005; Lyng et al., 2000; Mothersill & Seymour, 2002; Prise & O’Sullivan, 2009; Seymour & Mothersill, 2000; Smith et al., 2016). The cells that are in direct contact with each other are thought to support bystander signaling through the gap junctions (Prise & O’Sullivan, 2009). The experimental evidences also indicate that these effects are also mediated through the release of diffusive protein-like molecules, such as cytokines, from the cells that are irradiated or exposed to bystander signals. Recently, other factors such as exosomes containing RNA, UVA photons and NOS have been identified as potential candidates for bystander signals (Ahmad et al., 2013; Al-Mayah et al., 2012; Prise & O’Sullivan, 2009). The term “bystander signals” are generally used to denote multiple factors such as diffusive chemicals/factors (Fernandez-Palomo et al., 2015; Prise & O’Sullivan, 2009) or biophotons (Le et al., 2017), that contribute to the bystander effects in non-irradiated cells. Here, we will see a hybrid mathematical and computational modelling approach to study multiple effects of radiation and radiation-induced bystander effects on tumor and normal cells (Powathil et al., 2016). In this model, we consider bystander effects are produced only via bystander signals that diffuse through the medium/microenvironment (Fernandez-Palomo et al., 2015; Prise & O’Sullivan, 2009). In an in vitro study, it has been shown that these signals produced by the irradiated cells reach a maximum after 30 min of radiation and remain steady for at least 6 h after the radiation, transferring anywhere within the medium (Hu et al., 2006). The non-radiated cells that are exposed to these bystander signals respond with varying effects (Fig. 5.4).
The indirect radiation bystander effects are produced by radiation-induced signals sent by irradiated cells that are directly exposed to the radiation (Prise & O’Sullivan, 2009). The summary of the hybrid model is given in Fig. 5.2. Here, we are using a cellular potts model approach in a CompuCell3D framework to implement the hybrid multiscale model. The “bystander signals” are included in to the model as a field of bystander signal con- centration (Bs (x, t)) which by diffusing to nearby cells, produces bystander effects (Powathil et al., 2016). This concentration of bystander signals acts as a collection of multiple bystander diffusible signals that are observed experimentally. Following Powathil et al., 2016, the spatio-temporal evolution of these signals is modelled by a reaction-diffusion equation, given by:
Here, Bs (x,t) denotes the strength or concentration of the signal at position x and at time t, Ds is the diffusion coefficient of the signal (which is assumed to be constant), rs is the rate at which the signal is produced by an irradiated cell, cellRad (Ω, t) (cellRad (Ω, t) = 1 if position x ∈ Ω is occupied by a signal-producing irradiated cell at time t and zero otherwise) and ηs is the decay rate of the signal (Powathil et al., 2016).
We assume a heterogeneous distribution of cells where the cancer cells are surrounded by normal cells. Moreover, cancer cells are assumed to proliferate continuously (based on their internal cell-cycle ) while normal cells proliferate with the availability of free space (contact growth inhibition). To study the direct and indirect effects of radiation, we consider that the tumor cells are treated homogeneously to the prescribed dose per fraction and the surrounding normal cells receives lower doses with decreasing intensity from the tumor boundary. Two other scenarios of radiation exposure are given in Powathil et al. (2016).
The cells that are directly exposed to the radiation (including cells that undergo cell-death with LQ survival probability) are assumed to produce the bystander signals with some probability and these signals diffuse over the medium over time (4). Figure 5.5 shows an illustrative case of the spatio-temporal evolution of the cells (both normal and tumor cells) and corresponding signals when the cells are treated with one of 5 doses of 2 Gy radiation dose at time = 548 h. The light blue labelled cells in the upper panel of the Fig. 5.5 show the bystander signal producing cells that are under repair delay. The cells that are exposed to higher gradient of given dose are likely to undergo cell-death due to direct effects and cells that are exposed to low-dose radiation are likely to turn into a signal producing cells, resulting a higher number of signaling cells at low dose regions. The lower panel plots show the scaled signal concentrations at different time points with maximum value 3, beyond which bystander responses are activated (Powathil et al., 2016). The plots also show that, even if the level of bystander signals are minimal after each fraction, the contributions of bystander signals from multiple fractions can leads to a concentration level that might trigger bystander responses in responding cells. The parameters of the model are chosen in accordance with the experimental observations that the bystander signals are active at least up to around 6–10 h after radiation exposure (Hu et al., 2006).
The potential direct and indirect biological effects of radiation, and the effects that are incorporated into the model are illustrated in the Fig. 5.4. Since not all cells that are exposed to bystander signals respond to these signals (Gomez-Millan et al., 2012; Prise et al., 2005), it is assumed that the bystander responses are triggered probabilistically with higher chances for tumor cells than normal cells. Once the bystander signals are diffused within the medium, the non-targeted cells respond to these signals in multiple ways (bystander effects), depending on probability of each event to occur at a particular signal concentration level (Powathil et al., 2016). Depending on the signal concentration, these responses can be either protective or damaging (Fernandez-Palomo et al., 2015; Prise et al., 2005; Shuryak et al., 2007). The model considers both of these responses up to some extent and incorporate signal induced repair delay, signal induced bystander cell-kill and possibility of mutagenesis of normal cells. It is indeed hard to predict exact probabilities and concentration thresholds that are used within the model (Mothersill & Seymour, 2006). However, using sensitivity analysis, we have shown that qualitative behavior of the model remained unaffected with a relative change in the parameters. The details of this probabilistic model and the parameters that are used are given in Powathil et al. (2016).

Diagram on the left shows the multiple biological effects of radiation (left). Here, classical radiation biology operates within the area shaded green and bystander effects operate within the area shaded grey. Diagram on the right shows various interactions that are incorporated into the computational model when a growing tumor within normal tissue is irradiated. Here, we have added the responses of both normal and tumor cells. (Adapted from Powathil et al. (2016))

Plots showing the spatio-temporal evolution of host-tumor dynamics with and without treatment. Plots show the changes in the spatial distribution of irradiated cells and bystander signals when the host-tumor system is irradiated at times = 548 h. Upper panel shows the changes in cell distribution as well as the signaling cells after irradiation and the lower panel shows the distribution of bystander signals with color map indicating various threshold values. (Adapted from Powathil et al. (2016))

Plots show (a) the number of cells killed under the direct effects and indirect effects of radiation and other bystander signal responses, (b) the differences in the survival fraction when bystander responses are considered and (c) Experimental result: survival of asynchronous T98G human glioma cells irradiated with 240 kVp X-rays, measured using the cell-sort protocol (Figure from Joiner et al. (2001), used with copyright permission). (Adapted from Powathil et al. (2016))
Figure 5.6 shows the direct and indirect effects of radiation when the cells are exposed to 5 fractions of varying radiation doses (Powathil et al., 2016). The total cell-kill due to the exposed radiation and contributions from targeted and non-targeted cell-kill are given in Fig. 5.6a. As expected, plots show an increase in cell-kill with an increase in the dose per fraction. However, at the lower doses (dose = 0.25 Gy and dose = 0.5 Gy) the contributions from the radiation-induced bystander cell-kill predominate the direct cell-kill , indicating that bystander signals might play a role in radiation damage at low dose levels. The survival fraction of the cells after radiation, with and without bystander cell-kill are shown in Fig. 5.6b. The plots show a region on high cell-kill at the doses 0.25 Gy and 0.5 Gy as compared with doses greater than 0.5 Gy. The region of hyper radio-sensitivity at low dose levels or inverse-dose effect is also observed in several experimental studies, as shown in Fig. 5.6c (Joiner et al., 2001; Marples & Joiner, 2000; Prise et al., 2005) and is not predicted using the traditional LQ models. These in silico results were further supported by experimental study by Fernandez-Palomo et al. (2015), where they indicated that the bystander effects indeed play a role in hypersensitivity at low dose levels (Fernandez-Palomo et al., 2015; Powathil et al., 2016).
5 Conclusions and Overview
Over the past decade or so, with the rapid developments in acquisition of genetic, proteomic and other bio- chemical and biological data, systems biology has emerged as an important field in biomedical research. Systems biology aims to use interdisciplinary tools and skills to predict emergent behaviors in complex biomedical problems. Given the complex nature of most of the biomedical problems with inherent nonlinearities, included in cancer progression, systems biological approaches have been used recently to develop multiscale, multilevel predictive mathematical models to understand events that occur at multiple spatial and temporal level and resultant emergent behavior. Here, we discussed a hybrid multiscale approach that incorporates the details of intracellular, intercellular and cell-level to study the growth and progression of (cancer and/or normal) cells and its multiple direct and indirect effects to radiation. The multiscale model was implemented in two hybrid frameworks: hybrid multiscale CA framework and hybrid multiscale CPM framework (Brüningk et al., 2018; Powathil et al., 2012a; 2013, 2014). However, one should note that, in general, any predictions from (any) model is biologically relevant only when these assumptions are based on biological/clinical evidence and further, the results are validated with experimental data. Once validated using clinical and experimental data, these systems biological approaches, such as the ones described here, can provide mechanistic insights into radiation response and effectiveness, potentially helping to individualize treatment doses and multimodality treatment regimes (Brüningk et al., 2018; Powathil et al., 2013). As described in Sect. 5.3, we have validated the hybrid model to study the radiation effects in in vitro experiments to study fractionation protocols and multimodality treatments, in particular the combination with the heat (Brüningk et al., 2018).
The long-term aim of these modelling approaches are to develop qualitative, predictive mathematical and computational tools based on clinical and experimental data to understand, study, and provide useful predictions related to the outcome of multimodality treatment protocols used to treat human malignancies. This is an efficient and cost-effective alternative to test and study new treatment options, new dosage and efficacy of the treatments given, to allow the planning and designing of more adventurous treatments in silico, prior to beginning actual testing and long and costly clinical trials or experiments. As observed experimentally (Mothersill & Seymour, 2004; Munro, 2009; Prise & O’Sullivan, 2009), treatments such as radiation also induce indirect effects on the treated volume, that has a wider clinical implications in the treatment effectiveness and control, indicating the importance of studying and planning such protocols. One such indirect effect of the radiation is radiation induced bystander effect. Most of our knowledge about bystander effects is primarily from in vitro and in vivo studies, which may have less clinical applicability. Multiscale mathematical and computational tools that incorporate essential multi- layer complexities can be used as powerful tools to understand and identify the multiple parameters that are significant in radiation-induced bystander responses (Powathil et al., 2016). As described Sect. 5.4, such models can provide potential testable, qualitative hypotheses that may be very significant in exploring the role of bystander effects in a winder context. Our in silico findings about the role of bystander effects in mediating increased cell-kill at low doses were later supported by the experimental results by the Mothersill Group (Fernandez-Palomo et al., 2015).
Non-targeted and low dose effects of radiation have wider implications in an ecological perspective with a complex multilevel interaction among multiple species and their environment. Multiscale mathematical and computational modelling approaches can be extremely useful in studying the multilevel, long term effects/consequences of radiation (including the effects of historic doses) on a complex ecosystem in a qualitative approach, generating potentially testable hypothesis and inferences. The hybrid multiscale mathematical model outlined in this article to study the direct and indirect effects of radiation in the context of cancer progression serve as an example on how we can use mathematical modelling approaches to study complex biological problems. Moreover, it has many similarities in the multilevel spatial and temporal complexities involved in an ecosystem level. A step towards developing a predictive ecological model would be a continuous dialogue with the experts in radiation ecology to understand the relevant questions that needed to be studied and incorporated into the multiscale modelling approach.
References
Ahmad, S. B., McNeill, F. E., Byun, S. H., Prestwich, W. V., Mothersill, C., Seymour, C., Armstrong, A., & Fernandez, C. (2013). Ultra-violet light emission from HPV-G cells irradiated with low let radiation from (90)Y; consequences for radiation induced bystander effects. Dose Response, 11, 498–516.
Al-Mayah, A. H., Irons, S. L., Pink, R. C., Carter, D. R., & Kadhim, M. A. (2012). Possible role of exosomes containing RNA in mediating nontargeted effect of ionizing radiation. Radiation Research, 177(5), 539–545.
Andasari, V., Roper, R. T., Swat, M. H., & Chaplain, M. A. (2012). Integrating intracellular dynamics using CompuCell3D and Bionet- solver: Applications to multiscale modelling of cancer cell growth and invasion. PLoS One, 7(3), e33,726.
Anderson, A. R., & Chaplain, M. A. (1998). Continuous and discrete mathematical models of tumor-induced angiogenesis. Bulletin of Mathematical Biology, 60, 857–899.
Azzam, E. I., de Toledo, S. M., Waker, A. J., & Little, J. B. (2000). High and low fluences of alpha-particles induce a G1 checkpoint in human diploid fibroblasts. Cancer Research, 60(10), 2623–2631.
Blyth, B. J., & Sykes, P. J. (2011). Radiation-induced bystander effects: What are they, and how relevant are they to human radiation exposures? Radiation Research, 176(2), 139–157.
Brüningk, S., Powathil, G., Ziegenhein, P., Ijaz, J., Rivens, I., Nill, S., Chaplain, M., Oelfke, U., & ter Haar, G. (2018). Combining radiation with hyperthermia: a multiscale model informed by in vitro experiments. Journal of the Royal Society Interface, 15(138), 20170681
Fernandez-Palomo, C., Mothersill, C., Brauer-Krisch, E., Laissue, J., Seymour, C., & Schultke, E. (2015). H2AX as a marker for dose deposition in the brain of wistar rats after synchrotron microbeam radiation. PLoS One, 10(3), e0119,924.
Fowler, J. F. (1989). The linear-quadratic formula and progress in fractionated radiotherapy. British Journal of Radiology, 62(740), 679–694.
Franken, N. A. P., Rodermond, H. M., Stap, J., Haveman, J., & van Bree, C. (2006). Clonogenic assay of cells in vitro. Nature Protocols, 1(5), 2315–2319.
Glazier, J. A., Balter, A., & Popławski, N. J. (2007). Magnetization to morphogenesis: A brief history of the glazier-graner-hogeweg model. In Single-cell-based models in biology and medicine (pp. 79–106). Springer.
Gomez-Millan, J., Katz, I. S., Farias, V. D. E. A., Linares-Fernandez, J. L., Lopez-Penalver, J., Ortiz-Ferron, G., Ruiz-Ruiz, C., Oliver, F. J., & Ruiz de Almodovar, J. M. (2012). The importance of bystander effects in radiation therapy in melanoma skin-cancer cells and umbilical-cord stromal stem cells. Radiotherapy and Oncology, 102(3), 450–458.
Hu, B., Wu, L., Han, W., Zhang, L., Chen, S., Xu, A., Hei, T. K., & Yu, Z. (2006). The time and spatial effects of bystander response in mammalian cells induced by low dose radiation. Carcinogenesis, 27(2), 245–251.
Joiner, M. C., Marples, B., Lambin, P., Short, S. C., & Turesson, I. (2001). Low-dose hypersensitivity: Current status and possible mechanisms. International Journal of Radiation Oncology Biology Physics, 49(2), 379–389.
Kansal, A. R., Torquato, S., Harsh, G. R., IV, Chiocca, E. A., & Deisboeck, T. S. (2000). Cellular automaton of idealized brain tumor growth dynamics. Biosystems, 55, 119–127.
Lauber, K., Ernst, A., Orth, M., Herrmann, M., & Belka, C. (2012). Dying cell clearance and its impact on the outcome of tumor radiotherapy. Frontiers in Oncology, 2(9), 116.
Le, M., Fernandez-Palomo, C., McNeill, F. E., Seymour, C. B., Rainbow, A. J., & Mothersill, C. E. (2017). Exosomes are released by bystander cells exposed to radiation-induced biophoton signals: Reconciling the mechanisms mediating the bystander effect. PLoS One, 12(3), e0173, 685.
Lorimore, S. A., McIlrath, J. M., Coates, P. J., & Wright, E. G. (2005). Chromosomal instability in unirradiated hemopoietic cells resulting from a delayed in vivo bystander effect of gamma radiation. Cancer Research, 65(13), 5668–5673.
Lyng, F. M., Seymour, C. B., & Mothersill, C. (2000). Production of a signal by irradiated cells which leads to a response in unirradiated cells characteristic of initiation of apoptosis. British Journal of Cancer, 83(9), 1223–1230.
Marples, B., & Joiner, M. C. (2000). Modification of survival by DNA repair modifiers: A probable explanation for the phenomenon of increased radioresistance. International Journal of Radiation Biology, 76(3), 305–312.
Moreira, J., & Deutsch, A. (2002). Cellular automaton models of tumor development: A critical review. Advances in Complex Systems (ACS), 5(02), 247–267.
Morgan, W. F. (2003a). Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation-induced genomic instability and bystander effects in vitro. Radiation Research, 159(5), 567–580.
Morgan, W. F. (2003b). Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiation Research, 159(5), 581–596.
Mothersill, C., & Seymour, C. B. (2002). Bystander and delayed effects after fractionated radiation exposure. Radiation Research, 158(5), 626–633.
Mothersill, C., & Seymour, C. B. (2004). Radiation-induced bystander effects–implications for cancer. Nature Reviews in Cancer, 4(2), 158–164.
Mothersill, C., & Seymour, C. (2006). Radiation-induced bystander effects: Evidence for an adaptive response to low dose exposures? Dose Response, 4(4), 283–290.
Munro, A. J. (2009). Bystander effects and their implications for clinical radiotherapy. Journal of Radiological Protection, 29(2A), A133–A142.
Novak, B., & Tyson, J. J. (2003). Modelling the controls of the eukaryotic cell cycle. Biochemical Society Transactions, 31, 1526–1529.
Patel, A. A., Gawlinski, E. T., Lemieux, S. K., & Gatenby, R. A. (2001). A cellular automaton model of early tumor growth and invasion. Journal of Theoretical Biology, 213, 315–331.
Poleszczuk, J., & Enderling, H. (2014). A high-performance cellular automaton model of tumor growth with dynamically growing domains a high-performance cellular automaton model of tumor growth with dynamically growing domains. Applied Mathematics, 5(1), 144–152.
Powathil, G. G., Gordon, K. E., Hill, L. A., & Chaplain, M. A. (2012a). Modelling the effects of cell-cycle heterogeneity on the response of a solid tumour to chemotherapy: Biological insights from a hybrid multiscale cellular automaton model. Journal of Theoretical Biology, 308, 1–9.
Powathil, G., Kohandel, M., Milosevic, M., & Sivaloganathan, S. (2012b). Modeling the spatial distribution of chronic tumor hypoxia: Implications for experimental and clinical studies. Computational Mathematical Methods in Medicine, 2012, 410, 602.
Powathil, G. G., Adamson, D. J., & Chaplain, M. A. (2013). Towards predicting the response of a solid tumour to chemotherapy and radiotherapy treatments: Clinical insights from a computational model. PLoS Computational Biology, 9(7), e1003,120.
Powathil, G. G., Chaplain, M. A. J., & Swat, M. (2014). Investigating the development of chemotherapeutic drug resistance in cancer: A multiscale computational study. arXiv, 1407.0865 [q-bio.TO].
Powathil, G. G., Swat, M., & Chaplain, M. A. (2015). Systems oncology: Towards patient-specific treatment regimes informed by multiscale mathematical modelling. Seminars in Cancer Biology, 30, 13–20.
Powathil, G. G., Munro, A. J., Chaplain, M. A., & Swat, M. (2016). Bystander effects and their implications for clinical radiation therapy: Insights from multiscale in silico experiments. Journal of Theoretical Biology, 401, 1–14.
Prise, K. M., & O’Sullivan, J. M. (2009). Radiation-induced bystander signalling in cancer therapy. Nature Reviews in Cancer, 9(5), 351–360.
Prise, K. M., Schettino, G., Folkard, M., & Held, K. D. (2005). New insights on cell death from radiation exposure. Lancet Oncology, 6(7), 520–528.
Ribba, B., Colin, T., & Schnell, S. (2006). A multiscale mathematical model of cancer, and its use in analyzing irradiation therapies. Theoretical Biology and Medicine Modelling, 3, 7.
Richard, M., Kirkby, K., Webb, R., & Kirkby, N. (2007). A mathematical model of response of cells to radiation. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms, 255(1), 18–22.
Satelli, A., Annono, P., Azzinin, I., Campolongo, F., Ratto, M., & Tarantola, S. (2010). Varianance based sensitivity analysis of model output. Design and estimator for the total sensitivity index. Computer Physics Communications, 181(2), 259–270.
Seymour, C. B., & Mothersill, C. (2000). Relative contribution of bystander and targeted cell killing to the low-dose region of the radiation dose-response curve. Radiation Research, 153(5 Pt 1), 508–511.
Shuryak, I., Sachs, R. K., & Brenner, D. J. (2007). Biophysical models of radiation bystander effects: 1. Spatial effects in three- dimensional tissues. Radiation Research, 168(6), 741–749.
Smith, R. W., Seymour, C. B., Moccia, R. D., & Mothersill, C. E. (2016). Irradiation of rainbow trout at early life stages results in trans-generational effects including the induction of a bystander effect in non-irradiated fish. Environmental Research, 145, 26–38.
Turner, S., & Sherratt, J. A. (2002). Intercellular adhesion and cancer invasion: A discrete simulation using the extended Potts model. Journal of Theoretical Biology, 216, 85–100.
Tyson, J. J., & Novak, B. (2001). Regulation of the eukaryotic cell cycle: Molecular antagonism, hysteresis, and irreversible transitions. Journal of Theoretical Biology, 210, 249–263.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this paper

Cite this paper
Brüningk, S.C., Powathil, G.G. (2022). Modelling Direct and Indirect Effects of Radiation: Experimental, Clinical and Environmental Implications. In: Wood, M.D., Mothersill, C.E., Tsakanova, G., Cresswell, T., Woloschak, G.E. (eds) Biomarkers of Radiation in the Environment. NATO Science for Peace and Security Series A: Chemistry and Biology. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-2101-9_5
Download citation
DOI: https://doi.org/10.1007/978-94-024-2101-9_5
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-024-2100-2
Online ISBN: 978-94-024-2101-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)