
Abstract
Antisense oligonucleotides (AONs) are small synthetic molecules of therapeutic interest for a variety of human disease. Their ability to bind mRNA and affect its splicing gives AONs potential use for exon skipping therapies aimed at restoring the dystrophin transcript reading frame for Duchenne muscular dystrophy (DMD) patients. The neutrally charged phosphorodiamidate morpholino oligomers (PMOs) are a stable and relatively nontoxic AON modification. To assess exon skipping efficiency in vitro, it is important to deliver them to target cells. Here, we describe a method for the delivery of PMOs to myoblasts by electroporation. The described protocol for the Amaxa 4D X unit nucleofector system allows efficient processing of 16 samples in one nucleocuvette strip, aiding in high-throughput PMO efficacy screens.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Antisense oligonucleotides (AONs) are versatile, powerful tools for the potential treatment of a variety of diseases. AONs are short synthetic oligonucleotides consisting of modified DNA or RNA nucleic acid analogs. AONs can be exploited in multiple ways, including the modulation of splicing. Here, AONs bind to the unspliced mRNA and mask splice sites or exonic splice enhancer or silencer sites, resulting in an exon being ex- or included in the mature mRNA . Examples of such AONs are eteplirsen, golodirsen, viltolarsen, and casimersen to treat Duchenne muscular dystrophy (DMD) patients with eligible mutations, and nusinersen to treat spinal muscular atrophy (SMA). For DMD, antisense-mediated exon skipping is used to restore the reading frame of the dystrophin (DMD ) transcript , allowing the production of an internally deleted partially functional dystrophin protein [1]. This approach is mutation specific. Currently, for Duchenne, four AONs have been approved by the Food and Drug Administration (FDA, USA), which induce skipping of exon 51 (eteplirsen), exon 53 (golodirsen and viltolarsen) or exon 45 (casimersen).
Various different AON chemistries have been developed, such as 2′-O-methyl phosphorothioate (2′O-MePS), 2′-O-methoxyethyl phosphorothioate (2′-MOE-PS), and phosphorodiamidate morpholino oligomer (PMO) [2]. These different chemistries have unique chemical properties and were developed to enhance stability, solubility, and cellular uptake of AONs [2]. AONs are sometimes covalently conjugated to other molecules, in an attempt to improve their uptake by target tissues after systemic delivery [3]. For treatment of SMA, intrathecal injection of the AON into the cerebrospinal fluid (CSF) leads to efficient uptake by neurons and other cells in the nervous system with a long half-life [4]. However, in DMD all of the >700 different skeletal muscles are affected, and as such systemic delivery of AONs is required, which currently involves weekly intravenous infusions [5].
DMD consists of 79 exons, and there is a wide variety of unique patient mutations [6, 7], with a mutation hotspot spanning exon 45 through 53 [6, 8]. Approximately 55% of total DMD mutations causative for Duchenne would be eligible for some form of exon skipping therapy [7]. While skipping certain exons is applicable to larger groups of patients, it is crucial to skip also additional exons, which individually apply to small groups of patients, to increase the general applicability of this approach to as many patients as possible. To optimally design AONs for most of the DMD exons, it is important to have the ability for high-throughput screening of AON exon skipping efficacy. To perform reliable initial testing of AONs in vitro, it is essential to establish a reproducible, efficient means of delivery to a target cell. This can be achieved using electroporation of immortalized muscle cells [9]. Using immortalized cells has the advantage of theoretically unlimited proliferation, so large amounts of cells with homogenous characteristics can be generated. Primary cell sources are finite, and each new donor will have to be validated for reproducibility, which hampers screening potential when a large number of different AONs are to be tested simultaneously. Furthermore, it is our experience that the capacity of primary cultures to differentiate into myotubes declines with advanced passages.
Unlike 2′O-MePS AONs and dsDNA (e.g., plasmids), PMO AONs are neutrally charged, impeding the delivery by cationic lipid transfection systems. An alternative method for delivery of PMOs to mammalian cells is electroporation [9]. Electroporation relies on the formation of pores in the cell membrane by the application of an electric pulse through the transfection medium, mediating delivery of the particle of interest. The pore-forming pulse either serves to simultaneously deliver a charged molecule or is followed by a dedicated secondary delivery pulse. While electroporation efficiency is aided by active mobilization of charged molecules of interest into the cells by the electric current applied, the pore formation itself is already able to allow passive entry of inert molecules such as PMOs, albeit potentially at a lower efficiency. In fact, efficiency of in vitro PMO electroporation is relatively high when compared to other methods such as gymnosis and calcium-enriched medium (CEM) [10].
Classic electroporation is performed with cells in suspension, requiring dissociation of adherent cells from the culture vessel prior to the procedure. For studying DMD exon skipping, a cell line which expresses full-length DMD (Dp427m) is required. Alternatively, if available, a patient-derived cell line with a specific mutation can be used where skipping of a specific exon restores dystrophin production. However, as DMD expression in proliferating myoblasts is very low, myoblasts need to differentiate after electroporation with the Amaxa 4D X unit to form mature myotubes, expressing higher levels of DMD . Novel electroporation techniques also allow for electroporation of adherent cell layers, which can be advantageous when working with cells that grow slowly or have limited proliferation capacity upon differentiation, such as mature myotubes or neuronal cells. These methods, just like other electroporation techniques, require thorough characterization and optimization of conditions for maximum efficiency and are not included in this chapter.
In this chapter, we provide a protocol for the delivery of PMOs to immortalised myoblasts by electroporation with the Lonza Amaxa 4D-nucleofector X unit, and their subsequent differentiation to DMD expressing myotubes. The procedure for further sample processing and analysis of the skipped transcript by endpoint reverse transcription polymerase chain reaction (RT-PCR) and quantitative PCR (RT-qPCR) are also outlined.
2 Materials
2.1 Cell Culture
-
1.
Immortalized myoblasts: 0.5 × 106 up to 1 × 106 cells per reaction (for transfer to a 6-well plate) (see Note 1).
-
2.
Culture medium (proliferation): For myoblasts either: F10 Nutrient mix (nutmix) medium supplemented with 20% fetal bovine serum (FBS), 1% PenStrep, 10 ng/mL rhFGF and 1 mM dexamethasone, or Skeletal Muscle Cell Growth Medium (SMCGM) supplemented with 15% FBS and 50 μg/mL gentamicin (see Note 2).
-
3.
Culture medium (resuspension): F10 Nutmix + 20% FBS + 1% PenStrep.
-
4.
Culture medium (differentiation): DMEM (4.5 g/L glucose) + 2% FBS or 2% knockout serum replacement (KOSR) + 50 μg/mL Gentamicin or 1% PenStrep (see Note 2).
-
5.
Trypsin-EDTA (0.05%).
-
6.
Dulbecco’s phosphate-buffered saline (dPBS (-MgCl2, -CaCl2)).
-
7.
Culture vessels (e.g., T182 flask and 6-well plates).
2.2 Electroporation
-
1.
Lonza 4D nucleofector core unit.
-
2.
Lonza 4D nucleofector X-unit for cells in suspension (see Note 3).
-
3.
Suitable 4D-nucleofector X kit (e.g., primary cell optimization kit) containing 16-well nucleofector strips and nucleofection buffer.
-
4.
PMOs at 1 mM concentration in saline, 0.2-μm filter sterilized (see Notes 4 and 5).
2.3 RNA Isolation
-
1.
TRI-reagent (e.g., TRIsure).
-
2.
Chloroform.
-
3.
2-Propanol.
-
4.
70% Ethanol (EtOH).
-
5.
RNase-free water (DEPC treated).
2.4 cDNA Synthesis
-
1.
Random hexamer primers (N6) (20 ng/μL).
-
2.
dNTP mix (10 mM each).
-
3.
5× reverse transcriptase (RT) reaction buffer.
-
4.
Reverse transcriptase enzyme.
-
5.
RNase inhibitor.
-
6.
RNase-free water (DEPC treated).
2.5 RT-PCR Analysis of Skipping Efficiency
-
1.
cDNA generated from >1 μg total RNA by random hexamer primers (Subheading 3.3, step 5).
-
2.
Forward and reverse primer set (preferably intron spanning) (10 μM stock).
-
3.
dNTP mix (10 mM each).
-
4.
10× Reaction buffer.
-
5.
Taq polymerase.
-
6.
Molecular biology grade agarose.
-
7.
TRIS-Borate-EDTA (TBE) buffer (1×).
-
8.
Ethidium bromide (EtBr).
-
9.
100 bp DNA marker.
2.6 RT-qPCR of Skipping Efficiency
-
1.
cDNA generated from >1 μg total RNA by random hexamer primers (Subheading 3.3, step 5).
-
2.
Forward and reverse primer set for the gene of interest (one of the primers should span the exon boundary of the skipped product (e.g., the primer should consist of the last 10 nucleotides of exon 1, and the first 10 nucleotides of exon 3 of the gene of interest when skipping exon 2)) (10 μM stock).
-
3.
Forward and reverse primer set for a suitable reference gene (e.g., GUSB or GAPDH) (10 μM stock).
-
4.
2× SYBR-green PCR mastermix.
3 Methods
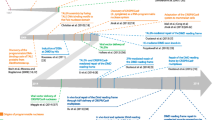
RNA isolated from the nucleofected myotube cultures is used to generate cDNA, which can subsequently be used for RT-PCR or RT-qPCR analysis of exon skipping efficiency. To purify a sufficient amount of total RNA from myotube cultures, we usually transfer cells to six-well plates after nucleofection. Smaller culture vessel might be suitable, or more optimal, for other purposes. We use up to 1 × 106 myoblast cells per 20 μL nucleofection reaction, requiring 1.6 × 107 cells in total for a full 16-well cuvette strip. A T182 culture vessel of high confluence contains about 2 × 107 myoblast cells. A graphical overview of the procedure is outlined in Fig. 1. All steps described in Subheading 3.1, steps 1–17 should be performed under aseptic conditions. Subheading 3.1, steps 18 through Subheading 3.2, step 11 should be performed using suitable personal protection according to local regulations.

Schematic workflow of nucleofecting PMO in myoblast cells
3.1 Nucleofection and Maintenance of Cells
This example assumes an experiment involving 1 full 16-well nucleocuvette strip, using 1 × 106 cells per reaction. Scale volumes up or down according to experimental design . Prior to starting the procedure, make sure that the nucleofector buffer has been supplemented as indicated. Prior to handling the cells, make sure that the desired programs to be used have been entered into the 4D-nucleofector core unit to avoid delays in the procedure when handling resuspended cells (see Note 6).
-
1.
Preferentially subculture myoblasts 1 day prior to nucleofection to ensure proper viability and growth phase of cells. Cells from confluent cultures might exhibit a decreased efficiency or survival.
-
2.
Label wells and add media to the culture vessels used to transfer cells post-nucleofection, e.g., 3 mL proliferation media per well of a 6-well plate. Place plates in a 37 °C CO2 incubator to warm and equilibrate the media.
-
3.
On the day of the nucleofection experiment, wash the cells with dPBS and trypsinize the cells. Use 2 mL trypsin 0.05% for a T182 and carefully tilt flask to cover all cells. Place cells with trypsin in a 37 °C incubator.
-
4.
When cells are properly detached from the surface (cell line dependent, but generally after 2 min), add resuspension media to a total volume of 10 mL to inhibit trypsin activity.
-
5.
Resuspend cells and transfer to a 50-mL tube (see Note 7). If multiple flasks are needed to obtain the required number of cells need for the experiment performed, pool cells at this step.
-
6.
Count the number of cells in the 50-mL tube (at least twice) using available cell counting methods. Transfer a total of 1.7 × 107 cells (16 1 × 106 reactions + 1 surplus) to a fresh conical tube, and pellet cells by centrifugation for 5 min at 200 relative centrifugal force (RCF).
-
7.
Aspirate medium completely and gently resuspend cell pellet in 20 μL nucleofection buffer per reaction (i.e., here we use 340 μL for 16 reactions + 1 surplus) (see Note 8).
-
8.
Aliquot 20 μL cell suspension in each of the chambers in the 16-well nucleofector strip using a P20 micropipette. Avoid creating air bubbles (see Note 9). When placing the cuvette strip with the yellow notch away from the user, the top left well is well A1 (see Note 10).
-
9.
Add 1 μL of PMO diluted to the desired concentration. For example, 1 μL PMO from a 1 mM stock will result in a final concentration in the cuvette of 50 μM. Gently mix by stirring with the pipet tip, do not vigorously pipet up and down (see Notes 11 and 12).
-
10.
Replace the lid on the cuvette strip (see Note 13) and place it in the 4D-Nucleofector X unit tray (yellow notch away from user, toward the machine).
-
11.
Start nucleofection by selecting start on the 4D-nucleofector core unit touch screen.
-
12.
After the machine has finished electroporating, check the status on the display. Green crosses mean the samples have been electroporated without issues. When other symbols are shown, an error might have occurred. Refer to the 4D-nucleofector user manual.
-
13.
Leave the nucleocuvette strip on the bench for 10 min for the cells to recover (see Note 14).
-
14.
Gently add 180 μL prewarmed media to each of the wells of the cuvette strip.
-
15.
Carefully resuspend (maximum 2–3 times up and down) the cells in the cuvette, and transfer to the culture vessel prepared in Subheading 3.1, step 2 (see Note 15).
-
16.
Allow cells to proliferate for 24 up to 72 h and monitor viability of electroporated cells (see Note 16). Let cells proliferate until reaching 100% confluency.
-
17.
Upon reaching confluency, replace myoblast proliferation media with differentiation media to induce myotube formation.
-
18.
After 72 h, wash cells with dPBS and lyse in 500 μL TRI-reagent (for a 6 well plate).
-
19.
Collect samples using a cell scraper and transfer to 1.5-mL microcentrifuge tubes on ice.
-
20.
Store samples (−80 °C) or immediately isolate total RNA from cells according to available protocols (e.g., 3.2).
3.2 RNA Isolation
-
1.
Thaw the lysate obtained at Subheading 3.2, step 17 and add 1/5th volume (100 μL) of chloroform (see Notes 17 and 18).
-
2.
Mix thoroughly by shaking for 30 s.
-
3.
Incubate on ice for 5 min.
-
4.
Centrifuge samples for 15 min at 16,000 RCF at 4 °C.
-
5.
Carefully transfer the upper aqueous phase to a new microcentrifuge tube, without disturbing the organic- and interphase.
-
6.
Add an equal volume of 2-propanol to transferred aqueous phase.
-
7.
Mix well and incubate for >30 min on ice, then centrifuge for 15 min at 16,000 RCF at 4 °C.
-
8.
Discard supernatant and wash pellet with 1 mL 70% EtOH.
-
9.
Centrifuge for 5 min at 16,000 RCF at 4 °C.
-
10.
Discard supernatant and air-dry pellet.
-
11.
Dissolve the pellet in RNase-free water (e.g., 25 μL).
-
12.
Determine RNA concentration using a Nanodrop ND-1000.
-
13.
Store RNA at −80 °C (see Note 19).
3.3 cDNA Synthesis
-
1.
To generate first-strand cDNA using random hexamer using at least 1 μg of total RNA in a 20 μL reaction. In a 12.5 μL reaction mix the following components (see Note 20):
-
(a)
X μL RNA for a total of 1 μg.
-
(b)
1 μL dNTP mix (10 mM each).
-
(c)
1 μL Random hexamer primers (N6) (20 ng/μL).
-
(d)
X μL RNase-free water up to 12.5 μL.
-
(a)
-
2.
Incubate reaction for 5 min at 65 °C. Chill on ice for 2 min.
-
3.
To each tube add:
-
(a)
4 μL 5× RT-reaction buffer.
-
(b)
1 μL Reverse transcriptase enzyme.
-
(c)
0.5 μL RNase inhibitor.
-
(a)
-
4.
Incubate at 42 °C for 1 h.
-
5.
Incubate at 85 °C for 5 min.
-
6.
Dilute the 20 μL cDNA reaction to a final volume of 100 μL with ultrapure MQ.
-
7.
Store cDNA at −20 °C.
3.4 RT-PCR Analysis
-
1.
Set up RT-PCR (25 μL reactions) according to the follow set-up per reaction (see Note 21):
-
(a)
2.5 μL 10× Reaction buffer.
-
(b)
1 μL Forward primer (10 μM).
-
(c)
1 μL Reverse primer (10 μM).
-
(d)
1 μL dNTP mix (10 mM each).
-
(e)
0.2 μL Taq polymerase (5 U/μL).
-
(f)
10 μL cDNA from Subheading 3.3, step 5.
-
(g)
9.3 μL ultrapure water (up to 25 μL).
-
(a)
-
2.
Gently mix and place samples in a thermal cycler using the following program:
-
(a)
1: 5 min, 95 °C—initial melt.
-
(b)
2: 30 s, 95 °C—melt.
-
(c)
3: 30 s, 60 °C—annealing (change for specific primers).
-
(d)
4: 40 s, 72 °C—extension (~30 s per kb).
-
(e)
5: Go to step 2, 34 additional times (35 cycles total).
-
(f)
6: 5 min, 72 °C—final extension.
-
(a)
-
3.
Analyze PCR samples by standard ethidium bromide (EtBr) agarose gel electrophoresis, using a 2% agarose gel (for products <1000 bp) in TBE buffer at 125 V (Fig. 2) (see Note 22). Alternatively, accurate analysis of signal intensity can be measured by use of, e.g., an Agilent 2100 Bioanalyzer (see Note 23).

Example of the result of RT-PCR analysis after successful nucleofection of a DMD exon 51 targeting PMO in KM155 myotubes using various programs of the 4D-nucleofector. (a) Agarose gel electrophoresis of endpoint RT-PCR products using primers specific for DMD exon 47 through 52. Skipping of exon 51 will lead to the production of a smaller PCR product as indicated. Different lanes consist of different experimental combination of nucleofection buffers and programs. (b) Approximate quantification of agarose gel shown in A by FIJI analysis . Ratios of Exon 51 skipped product intensities over the regular (exon 51 containing) product are plotted. The average of the negative controls (no PMO/no pulse samples (orange bars)) is shown as a dotted line. (c) RT-qPCR analysis of DMD exon 51 skipping in KM155 myotubes with 6 nucleofection programs and 5 nucleofection buffers. Exon skipping was determined with a primer set only amplifying the DMD transcript without exon 51 (Exon 50-52F + Exon 52R). A primer set specific for DMD exon 49 through 50 shows all DMD transcript , and MYH3 is used as a marker for myogenic differentiation. Cells resuspended in nucleofection buffer not subjected to an electric pulse were used as a negative control. In our hands, exon skipping efficiency was highest using a combination of program CM-137 and buffer P1
3.5 RT-qPCR Analysis
-
1.
For RT-qPCR analysis , measure the gene of interest and at least one reference gene. Always measure a technical triplicate of each cDNA-primer combination. Set up RT-qPCR according to the following set-up per reaction (we use 8 μL reactions in a 384-well plate):
-
(a)
4 μL 2× SYB green master mix.
-
(b)
1 μL Forward primer (10 μM).
-
(c)
1 μL Reverse primer (10 μM).
-
(d)
2 μL cDNA from Subheading 3.3, step 5.
-
(a)
-
2.
Seal the plate and mix by inversion and briefly spinning down the plate in a centrifuge.
-
3.
Place samples in a real-time PCR enabled thermal cycler and run the following program:
-
(a)
1: 5 min, 95 °C—initial melt.
-
(b)
2: 10 s, 95 °C—melt.
-
(c)
3: 30 s, 60 °C—Annealing and extension (change temperature for specific primers).
-
(d)
4: Read plate, go to step 2, 39 additional times (40 cycles total).
-
(e)
5: Melt curve analysis .
-
(a)
-
4.
A suitable primer pair for the skipped product will yield little to no signal in the un-nucleofected control samples and will have increased abundance when the exon was successfully skipped.
4 Notes
-
1.
The procedure described herein has been optimized and validated using immortal muscle cell line KM155, which has been described previously [11, 12]. Use of this protocol for primary muscle cells or other immortalized cell lines might require additional optimization.
-
2.
Different media are described in the literature for proliferation and/or differentiation of myoblasts. These different media can have considerable effects on gene expression and morphology. For example, differentiation of myoblasts with 2% FCS leads to slower formation of multinucleated myotubes compared to 2% KOSR. However, myotubes formed with 2% KOSR are harder to handle due to faster release from the vessel surface, presumable due to spontaneous contraction of the myotubes. We recommend that different media are tested to compare optimal experimental conditions.
-
3.
For electroporation of adherent cells, a few of the options available are the Lonza 4D nucleofector Y-unit or the Nepagene NEPA21 electroporator.
-
4.
Delivery of PMOs by nucleofection can be prohibitively expensive for many laboratories, as prices for consumables are steep compared to common laboratory transfection reagents, some of which can be cheaply prepared in-house. On the other hand, if the material to be transfected is expensive or rare, nucleofection allows for relatively high concentrations to be applied directly to the cells due to the low reaction volume (as low as 20 μL), saving costs on oligonucleotides.
-
5.
While small molecules such as siRNAs and AONs are easily delivered into myoblasts by electroporation (e.g., siRNA , PMOs) or transfection (e.g., siRNA , 2OMePS AONs), delivery of (large) plasmids is notoriously inefficient.
-
6.
For our immortalized myoblast cell line (KM155), we have tested various different nucleofector pulse programs in combination with the nucleofection buffers present in the primary cell line optimization kit (i.e., P1, P2, P3, P4, and P5). We noticed that the trend of DMD exon skipping efficiency was similar for each buffer and depended largely on the nucleofection program used for the 4D X unit (Fig. 2c, bars). Buffers did however largely contribute to overall efficiency of a set of nucleofections (Fig. 2c, individual data points per bar).
-
7.
When trypsinizing cells from the culture vessel, dissociate cells by controlled but forceful pipetting with a 10-mL pipet and a pipetboy to generate a single-cell suspension. Cell clumps will severely hamper nucleofection.
-
8.
Cells should not be kept in nucleofection buffer for a prolonged amount of time, as this can reduce efficiency and viability. After diluting the cell pellet in nucleofection buffer, working swiftly and accurately is key.
-
9.
At any step of the procedure, but especially upon loading of the nucleocuvette, avoid air bubbles in the reagents containing cells. Cavitation shearing and electric arcing can occur when air bubbles are present. Loading the cuvette with a P20 and reverse pipetting (i.e., not pressing the piston to the second stop) are useful for avoiding air bubbles in the cuvette strip.
-
10.
We have tested the re-use of single cuvette strips for the delivery of the same PMO without any noticeable effect on delivery efficiency or cell viability. If the proprietary nucleofector solutions have not finished upon finishing the cuvette strips of a kit, this method can prolong the use of a single ordered kit. To clean a cuvette strip: after transferring cells from a nucleofection experiment from the 16-well strip to a culture vessel, immediately submerge the strip (without lid) in a-100 mL glass bottle filled with sterile milli-Q (MQ). Shake vigorously for 30 s. Decant MQ and add 70% EtOH for disinfection and shake vigorously. Do not leave the cuvette strip in ethanol for extended periods of time to prevent damage to the electrode. Decant EtOH and rinse twice with sterile MQ, airdry strip and store for future use. We have not tested degradation of efficiency after more than two re-uses. Different nucleofection programs with higher voltages might affect electrode degradation and result in poor efficiency upon re-use. Re-usability should always be tested in the experimental setup used to avoid problems in reproducibility.
-
11.
As an alternative to loading the PMO directly in the cell suspension, it is possible to mix PMO, cells and nucleofection buffer prior to dispensing in the 16-wells nucleocuvette. For example, in bulk when multiple pulse programs are tested, or in sterile strips/plates for testing large amounts of different PMOs. For the latter, it is possible to transfer cells to the nucleocuvette by use of a multichannel pipette.
-
12.
It is possible to add the PMO to the nucleocuvette wells prior to adding the cells, in which addition of the cells and handling of the strip will sufficiently mix the substrate with the cell suspension. However, as PMO solutions tend to be slightly viscous, it can be hard to reliably dispense a small volume (e.g., 1 or 2 μL) in a dry well, compared to dispensing in solution.
-
13.
The cell suspension (20 μL) in the nucleocuvette strip should cover the entire bottom of the well in the nucleocuvette. After loading the cell suspension and prior to placing the cuvette in the X-unit, tap it several times on the working surface with appropriate force.
-
14.
Excessive cell death may warrant changes in recovery steps. For example, after electroporation, add 180 μL warm culture media to each well of the cuvette strip, and incubate the strip 10 min at 37 °C. Afterwards, transfer cells carefully as described above.
-
15.
Cells are fragile immediately after electroporation. Avoid shear stress by over-resuspension with small diameter tip orifices. Use a P200 or P1000 micropipette, resuspend two or three times in the cuvette, and immediately transfer to the prewarmed culture plate. Avoid creation of air bubbles by aggressive pipetting.
-
16.
Certain cell types might show increased viability after nucleofection when treated with ROCK inhibitors to block apoptosis 4 h prior and 24 h post nucleofection.
-
17.
RNA is extremely sensitive to degradation by environmental RNases, such as present on human skin. When handling/preparing RNA samples, always wear suitable gloves, and clean work surfaces with RNase removal agents (e.g., RNaseZAP).
-
18.
To compare samples properly, it is important to process sample sets in the same way. This includes but is not limited to simultaneous isolation of RNA, simultaneous generation of cDNA, identical input of RNA for cDNA reactions, etc.
-
19.
For assessing the efficiency of DMD exon skipping with RT-PCR, we generally do not incorporate a DNase I treatment step in the RNA isolation protocol.
-
20.
Generation of cDNA can be performed in PCR strips with caps, allowing for easy incubations in a thermal cycler and transfer of sample to another PCR strip with a multichannel pipette.
-
21.
For analysis of exon skipping efficiency by RT-PCR, we strongly recommend the use of single-reaction RT-PCR instead of nested PCR amplifications. Nested PCR amplifications have a high tendency to induce preferential amplification bias and are less quantitatively reliable [13]. Different polymerases and protocols might be better suited for specific transcripts and should be tested.
-
22.
When using software such as ImageJ/FIJI to analyze agarose gel band intensities, it is of the utmost importance that the user keeps the limitations of the software in mind. Saturated signal on the source image will lead to misleading results if analyzed incorrectly. Therefore, while useful for estimating relative intensity, many applications will require more sensitive methods to reliably quantify exon skipping efficiency. Furthermore, detection of DNA in agarose gels is facilitated by the amount of EtBr intercalating with the DNA, resulting in signal when exposed to UV light. As shorter PCR products bind less EtBr, their intensity is inherently underestimated when measured in analysis software.
-
23.
Prior to analyzing PCR products on an Agilent 2100 bioanalyzer, it is advised to always run some of the sample on an EtBr agarose gel to confirm successful PCR amplification.
References
Aartsma-Rus A, van Ommen GJ (2007) Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA 13(10):1609–1624. https://doi.org/10.1261/rna.653607
Douglas AG, Wood MJ (2013) Splicing therapy for neuromuscular disease. Mol Cell Neurosci 56:169–185. https://doi.org/10.1016/j.mcn.2013.04.005
Jirka SM, Heemskerk H, Tanganyika-de Winter CL, Muilwijk D, Pang KH, de Visser PC, Janson A, Karnaoukh TG, Vermue R, t Hoen PA, van Deutekom JC, Aguilera B, Aartsma-Rus A (2014) Peptide conjugation of 2′-O-methyl phosphorothioate antisense oligonucleotides enhances cardiac uptake and exon skipping in mdx mice. Nucleic Acid Ther 24(1):25–36. https://doi.org/10.1089/nat.2013.0448
Neil EE, Bisaccia EK (2019) Nusinersen: a novel antisense oligonucleotide for the treatment of spinal muscular atrophy. J Pediatric Pharmacol Ther 24(3):194–203. https://doi.org/10.5863/1551-6776-24.3.194
Aartsma-Rus A, Krieg AM (2017) FDA approves Eteplirsen for Duchenne muscular dystrophy: the next chapter in the Eteplirsen saga. Nucleic Acid Ther 27(1):1–3. https://doi.org/10.1089/nat.2016.0657
Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT (2006) Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34(2):135–144. https://doi.org/10.1002/mus.20586
Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Roy AJ, Chamova T, Guergueltcheva V, Chan S, Korngut L, Campbell C, Dai Y, Wang J, Barišić N, Brabec P, Lahdetie J, Walter MC, Schreiber-Katz O, Karcagi V, Garami M, Viswanathan V, Bayat F, Buccella F, Kimura E, Koeks Z, van den Bergen JC, Rodrigues M, Roxburgh R, Lusakowska A, Kostera-Pruszczyk A, Zimowski J, Santos R, Neagu E, Artemieva S, Rasic VM, Vojinovic D, Posada M, Bloetzer C, Jeannet PY, Joncourt F, Díaz-Manera J, Gallardo E, Karaduman AA, Topaloğlu H, El Sherif R, Stringer A, Shatillo AV, Martin AS, Peay HL, Bellgard MI, Kirschner J, Flanigan KM, Straub V, Bushby K, Verschuuren J, Aartsma-Rus A, Béroud C, Lochmüller H (2015) The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 36(4):395–402. https://doi.org/10.1002/humu.22758
Tuffery-Giraud S, Béroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, Moizard MP, Bernard R, Cossée M, Boisseau P, Blayau M, Creveaux I, Guiochon-Mantel A, de Martinville B, Philippe C, Monnier N, Bieth E, Khau Van Kien P, Desmet FO, Humbertclaude V, Kaplan JC, Chelly J, Claustres M (2009) Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat 30(6):934–945. https://doi.org/10.1002/humu.20976
Aung-Htut MT, McIntosh CS, West KA, Fletcher S, Wilton SD (2019) In vitro validation of phosphorodiamidate morpholino oligomers. Molecules 24(16):2922. https://doi.org/10.3390/molecules24162922
Hori S-I, Yamamoto T, Waki R, Wada S, Wada F, Noda M, Obika S (2015) Ca2+ enrichment in culture medium potentiates effect of oligonucleotides. Nucleic Acids Res 43(19):e128. https://doi.org/10.1093/nar/gkv626
Mamchaoui K, Trollet C, Bigot A, Negroni E, Chaouch S, Wolff A, Kandalla PK, Marie S, Di Santo J, St Guily JL, Muntoni F, Kim J, Philippi S, Spuler S, Levy N, Blumen SC, Voit T, Wright WE, Aamiri A, Butler-Browne G, Mouly V (2011) Immortalized pathological human myoblasts: towards a universal tool for the study of neuromuscular disorders. Skelet Muscle 1:34. https://doi.org/10.1186/2044-5040-1-34
Echigoya Y, Lim KRQ, Trieu N, Bao B, Miskew Nichols B, Vila MC, Novak JS, Hara Y, Lee J, Touznik A, Mamchaoui K, Aoki Y, Takeda S, Nagaraju K, Mouly V, Maruyama R, Duddy W, Yokota T (2017) Quantitative antisense screening and optimization for exon 51 skipping in Duchenne muscular dystrophy. Mol Ther 25(11):2561–2572. https://doi.org/10.1016/j.ymthe.2017.07.014
Spitali P, Heemskerk H, Vossen RHAM, Ferlini A, den Dunnen JT, t Hoen PAC, Aartsma-Rus A (2010) Accurate quantification of dystrophin mRNA and exon skipping levels in duchenne muscular dystrophy. Lab Investig 90(9):1396–1402. https://doi.org/10.1038/labinvest.2010.98
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Goossens, R., Aartsma-Rus, A. (2022). In Vitro Delivery of PMOs in Myoblasts by Electroporation. In: Arechavala-Gomeza, V., Garanto, A. (eds) Antisense RNA Design, Delivery, and Analysis. Methods in Molecular Biology, vol 2434. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2010-6_12
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2010-6_12
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2009-0
Online ISBN: 978-1-0716-2010-6
eBook Packages: Springer Protocols