
Abstract
The possible evolutionary trajectories a population can follow is determined by the fitness effects of new mutations. Their relative frequencies are best specified through a distribution of fitness effects (DFE) that spans deleterious, neutral, and beneficial mutations. As such, the DFE is key to several aspects of the evolution of a population, and particularly the rate of adaptive molecular evolution (α). Inference of DFE from patterns of polymorphism and divergence has been a longstanding goal of evolutionary genetics.
polyDFE provides a flexible statistical framework to estimate the DFE and α from site frequency spectrum (SFS) data. Several probability distributions can be fitted to the data to model the DFE. The method also jointly estimates a series of nuisance parameters that model the effect of unknown demography as well data imperfections, in particular possible errors in polarizing SNPs. This chapter is organized as a tutorial for polyDFE. We start by briefly reviewing the concept of DFE, α, and the principles underlying the method, and then provide an example using central chimpanzees data (Tataru et al., Genetics 207(3):1103–1119, 2017; Bataillon et al., Genome Biol Evol 7(4):1122–1132, 2015) to guide the user through the different steps of an analysis: formatting the data as input to polyDFE, fitting different models, obtaining estimates of parameters uncertainty and performing statistical tests, as well as model averaging procedures to obtain robust estimates of model parameters.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Distribution of fitness effects
- Rate of adaptive molecular evolution
- Beneficial mutations
- Polymorphism and divergence data
1 Introduction
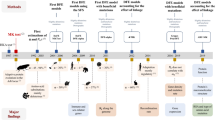
The following tutorial requires the successful installation of polyDFE-v1.1 (see manual for details on installation), and basic skills in using the command line and R. The latest version of polyDFE, its manual as well as an R script postprocessing.R that contains functions which facilitate post-processing of polyDFE output files can be found on https://github.com/paula-tataru/polyDFE.
1.1 Modelling the Properties of Mutations on Fitness
Genome and exome sequencing studies open the possibility to survey systematically nucleotide variation in genomes. Several model-based methods have been developed to infer the properties of mutations from these surveys. In a nutshell, population genetics models introduced in Chapter 1 can formalize a fundamental intuition: the fitness effect of a new mutation will influence the frequency at which it segregates in a population. More formally, assuming a set of independent SNPs, mathematical expectations can be obtained that relate mutation rates and the fitness effect of mutations to observable quantities such as the number of SNPs that are found at a given frequency (i.e., the counts of the site frequency spectrum, SFS) in a sample of individuals that were re-sequenced or genotyped [1].
The effects of new mutations on fitness are expected to vary depending on the region where the mutation happens and what types of changes are incurred by the mutation. We model the variation in effects of mutations by making a number of assumptions:
-
We can make an a priori distinction between sites where only neutrally evolving mutations are segregating and sites that harbor mutations potentially under selection. We refer to these as neutral/selected sites, respectively.
-
The number of mutations in a region of known length of nucleotides arises randomly as a Poisson process with a certain intensity that depends on the length of the region and the mutation rate per nucleotide.
-
Mutations that happen at neutral sites are lost or drift to fixation solely due to genetic drift.
-
Mutations happening at selected sites are ascribed a fitness effect through a scaled selection coefficient. Each mutation at a selected site is treated as exchangeable: no sites are identified a priori as yielding mutations that are intrinsically good or bad for fitness. The scaled selection coefficient 4Nes of a mutation is drawn at random from an underlying distribution, also called the distribution of fitness effects (DFE).
polyDFE performs maximum likelihood (ML) inference of DFE parameters from polymorphism data. Various probability distributions have been used to model DFEs [2]. Currently, polyDFE uses four types of distributions, referenced as models A through D and described in detail in the polyDFE manual. In this chapter, we focus solely on examples where models A and C are used.
Under model A, the DFE is given by a reflected and displaced Γ distribution. This distribution is parameterized through a mean scaled selection coefficient \( \overline{S} \), a shape b, and a maximum scaled selection coefficient Smax. This continuous distribution is theoretically motivated as the approximation for the DFE expected under an explicit fitness landscape where fitness is determined by k traits under stabilizing selection and where each mutation will pleiotropically affect every trait [2]. In the general case where Smax is not restricted to be 0, the DFE will comprise some beneficial mutations, otherwise the DFE will only comprise deleterious mutations.
Under model C, the DFE is given by a mixture of two distributions. A proportion pb of mutations are favorable and their scaled selection coefficient is drawn from an exponential distribution with mean Sb, and the remaining 1 − pb are deleterious mutations with scaled selection coefficient drawn from a reflected Γ distribution with mean Sd and shape b. If the proportion pb is restricted to be 0, the DFE will only comprise deleterious mutations. Note that the DFEs containing only deleterious mutations obtained from either model A (Smax = 0) or model C (pb = 0) are equivalent and are given by a reflected Γ distribution.
When inferring DFEs from SFS data, a DFE with only deleterious mutations (henceforth a deleterious DFE) is typically assumed. In order to obtain information about the selection coefficients of beneficial mutations, available methods rely on the amount of divergence data between the species of interest (ingroup) and an outgroup. polyDFE departs from this approach as it allows the user to obtain estimates of a DFE also containing beneficial mutations (henceforth a full DFE) solely from SFS data. In doing so, polyDFE has the advantage of not assuming that the DFE is a constant in both the ingroup and outgroup. The price to pay for relaxing this assumption is that by only using the SFS, the ML estimates have more sampling variance, reflecting the uncertainty due to reduced amounts of data.
1.2 Calculating the Rate of Adaptive Evolution, α
Obtaining estimates of the DFE allows one to learn more about factors governing the rate of adaptive molecular evolution, commonly defined as the proportion of fixed adaptive mutations, α. Besides ML estimates of DFE, polyDFE can be used to obtain ML estimates of α. Once the DFE is estimated, α can be obtained using the divergence data as:
where the expected number of neutral and deleterious substitutions is obtained from the DFE [1].
Alternatively, α can be estimated without using the divergence data by replacing the observed divergence selected counts with expectations derived from the DFE [1]. The two different estimations of α are referred to αdiv and αdfe [1], to reflect the type of data/information used.
Note that alternative statistics exist for measuring molecular adaptation, such as ωA, the rate of adaptive evolution relative to the mutation rate, or \( {K}_{a^{+}} \), the rate of adaptive amino acid substitutions [3, 4]. These have different properties and might be better suited for studying various aspects of adaptation [4]. Currently, polyDFE only calculates α, but these statistics can also be obtained once the DFE is estimated.
2 Pre-processing of the Data
2.1 The Type of Information Required by polyDFE
polyDFE requires as input the derived SFS at both neutral and selected sites. The input file can optionally also contain counts of divergence.
When preparing the data, there are three elements that require some careful attention:
-
what is the length of the region that was called for the potential occurrence of SNPs;
-
how are SNPs polarized into an ancestral and a derived allele;
-
how missing data is removed.
Software that enables SNP calling will also report the length of the region, calculated as the number of nucleotide positions where SNP calling could be performed. This length has to be then (correctly) divided between the data containing neutral sites and a priori selected sites. For an example, see the end of Subheading 2.2.
polyDFE assumes that the SFS is derived (polarized) and the given counts (see Subheading 2.2) are for derived SNP alleles. Various methods are available to orient SNPs, including parsimony and more rigorous probabilistic methods [5,6,7]. All methods require access to at least one outgroup.
polyDFE cannot deal with missing data. If the SFS contains missing data, several strategies can be used. If local linkage disequilibrium is known, SNP imputation can be used to estimate the missing genotypes. Alternatively, projection methods can be used to down-sample the SNP data to build a complete SFS with a reduced number of samples [8, 9].
2.2 Example of a polyDFE Input File
This tutorial uses central chimpanzee data [1, 10] to exemplify the different steps of an analysis. The central chimpanzee data and all additional files used here can be found on https://github.com/StatisticalPopulationGenomics. Here is a snippet of the chimpanzee data found in the input file central_chimp_sfs:

The first non-empty non-comment line specifies sequentially that there is only one (1) neutral and one (1) selected region and that 24 haplotypes were re-sequenced to obtain the SFS data.
Note that polyDFE can, in principle, analyze jointly multiple regions with different mutation rates that share the DFE parameters [1]. For the remainder of this chapter, we analyze data pooled into a single SFS for neutral and selected sites. For details on variability of mutation rates, see the polyDFE manual.
For each region (here two in total), the neutral followed by the selected ones, there is one line of input that gives sequentially the entries of the SFS, i.e. how many SNPs had the derived allele in 1 copy (14492 for the neutral region and 12645 for the selected region), 2 copies (6138 for the neutral region and 4573 for the selected region), up to 23 copies (845 for the neutral region and 469 for the selected region), followed by the length of the region (4292115 for the neutral region and 16146528 for the selected region), the divergence counts for the same region (44048 for the neutral region and 26481 for the selected region) and the length of the region where divergence data was obtained (4290192 for the neutral region and 16139295 for the selected region). The presence of divergence data in the input file is optional. For further details on the data format, see the polyDFE manual.
The central chimpanzee data was obtained from exome sequencing. To divide it into a neutrally evolving region and one potentially containing sites under selection, SNPs have been classified into synonymous and non-synonymous, with the first class assumed to be neutral [10]. This is a general practice when working with exome data. The length of the neutral (here, synonymous) and selected (here, non-synonymous) regions is typically calculated from the total length by using a proportionality principle where a proportion of the sites are deemed, respectively, synonymous and non-synonymous. Roughly we expect 3/4 of the sites in exome regions to be non-synonymous, but there are more rigorous and precise ways to calculate this quantity [11].
2.3 Note on SFS Data
A priori, one expects an L or U shaped SFS, where a lot of derived alleles are present in low frequencies and possibly a few more have high frequencies, especially when beneficial mutations contribute substantially to the SFS. This is borne out of population genetics theory and the fact that we expect that at selected sites a substantial fraction of the variation is deleterious to some degree. Large counts in intermediate frequencies are only caused by a significant amount of balancing selection or by cryptic and highly pronounced genetic differentiation in the sample of sequenced individuals.
3 Model Fitting with polyDFE
3.1 Specifying a DFE Model to Fit Using polyDFE
We use a series of examples to illustrate how to run polyDFE, and how to specify two key things:
-
the input file containing the data to be analyzed;
-
the DFE model used for the analysis.
The aim of the analysis is to fit a series of models differing by:
-
the type of DFE assumed;
-
the presence or not of beneficial mutations in the DFE;
-
the inclusion of two types of nuisance parameters that apply to both neutral and selected sites:
-
a polarization error εan;
-
a series of nuisance parameters ri, one for each class of frequency in the SFS.
-
The polarization error εan accounts for the fact that methods for orienting the SNPs are not perfect and still leave errors in the data, where the inferred derived allele is, in fact, the ancestral allele. When εan is set and fixed to 0, it is assumed that the data contains no error. To the best of our knowledge, polyDFE is the only available method that explicitly incorporates polarization error.
The nuisance parameters ri describe how the SFS can be distorted by sampling, demography, and/or linkage, relative to what is expected in a stable Wright-Fisher population at mutation-selection-drift equilibrium. When these parameters are fixed to 1, it is assumed that the data does not depart from standard expectations. For some datasets, errors in the SNP orientation can be efficiently captured by the distortion parameters ri [1, 12], but this is not always the case [1]. We always recommend inferring a full model where all parameters are estimated and use hypothesis testing to decide whether such additional parameters are needed or not (see Subheading 5.2).To simply run polyDFE on the chimpanzee data using default settings, the following command line can be used:

where
-
-d central_chimp_sfs specifies that polyDFE runs on the input file central_chimp_sfs.
However, it is more useful to customize the behavior of polyDFE through the command line arguments by specifying, for example, which DFE model is used or which parameters should be estimated or not:

where
-
-m A specifies that model A will be used to infer the DFE.
-
-i init_A.txt 1 specifies that the parameter configuration with ID 1 from the initialization file init_A.txt should be used. The initialization file is used to control which parameters should polyDFE estimate and to provide their initial values used during the estimation process. For example, polyDFE can be forced to estimate a deleterious DFE. The init_A.txt file provides examples on how parameters are set to be fixed or estimated.
-
-e specifies that the parameters’ initial values should be estimated automatically, using a combination of approximate analytical results and a grid search. Using -e is highly recommended when running an initial analysis.
-
-b params_basinhop.txt 1 specifies polyDFE to run a more involved likelihood maximization, see Subheading 3.2 for details.
-
>central_chimp_A (the redirection command) specifies that the output of polyDFE should be stored in the file central_chimp_A.
To run a full analysis on a dataset, models with increasing complexity (i.e. deleterious or full DFE, and allowing or not for nuisance parameters) should be used, as specified using -i. The example file run_polyDFE.txt contains all the command lines that allow a full analysis of the example dataset using both estimation under models A and C. As the true shape of the DFE is not known (i.e., is the DFE in the form of an A or C model?), it is recommended to run polyDFE with multiple models and finally use hypothesis testing to find the best fitting model (see Subheading 5.2).
If the input data also contains divergence counts, polyDFE uses this information for estimation by default. To use exclusively the SFS data during the estimation process, the command line argument -w is used.
3.2 Note on Likelihood Maximization
One of the key issues in obtaining reliable ML estimates is to ensure that the likelihood function is properly maximized over the space of parameters. This is not trivial, and besides the limitations of the method itself, this is what in practice will cause polyDFE to return poor estimates.
polyDFE implements multiple steps to ensure, as much as possible, that good estimates are found.
-
1.
The maximization algorithm requires initial values for the parameters, and these values can have a big effect on the estimates found. They can be provided by the user in the initialization file given through the command line argument -i, but for an initial run of polyDFE, it is strongly recommended to use the command line argument -e. This ensures that the maximization is started with sensible parameter values.
-
2.
Standard maximization algorithms allow parameters to take any value over the whole real line. Many of the parameters polyDFE estimates are constrained within a specific range. For example, the shape b for the DFE distribution is constrained to be positive. To allow for such constraints, polyDFE transforms the parameters from a given range to the whole real line. The range of each parameter can be controlled through the command line argument -r. polyDFE uses large ranges by default, but providing a range that is tighter around the potential maximum could improve the maximization process. For more details, see the polyDFE manual.
-
3.
Standard maximization algorithms, including the ones used in polyDFE, only ensure that a local maximum likelihood is found. However, a better solution can possibly exist. To avoid that polyDFE is stuck in a local solution, the basin hopping algorithm [13] can be used. This is a stochastic algorithm that runs the standard maximization algorithm multiple times, using different initial values for the parameters. polyDFE runs basin hopping when the command line argument -b is provided, as in the previous example. The basin hopping algorithm can be customized through a parameterization file. In the previous example, we used -b params_basinhop.txt 1, which specified that polyDFE should use the parameterization found in params_basinhop.txt with ID 1. The params_basinhop.txt file provides examples on how the basin hopping algorithm can be customized. For more details, see the polyDFE manual.
polyDFE uses maximization algorithms that rely on the gradient of the likelihood. If a set of parameters truly has a locally maximum likelihood, then the corresponding gradient is 0. If the gradient of the best solution found is far away from 0, this is warning sign that a good solution was not found. For different ways to change the run of polyDFE to aim for a better gradient, see the polyDFE manual.
4 Post-Processing of the polyDFE Output
polyDFE is accompanied by an R script postprocessing.R contains a series of functions written that parse the output of polyDFE and calculate other quantities of interest. We provide examples of polyDFE output files, as well as how to use the R functions to parse the output and perform hypothesis testing/model averaging on DFE and α. The analysis found below is also provided as an example file analysis.R.
4.1 Example of a polyDFE Output File
The output starts with a summary of how polyDFE was run, the progression of the maximization procedure, the best estimates found, various expectations under the best estimates (such as, the expected SFS) and, finally, estimates of α.
Here is a snippet of the output file central_chimp_A created in the previous example:

For more details on the misattributed polymorphism, see Subheading 4.4.
4.2 Merging and Parsing Output Files
The R functions from postprocessing.R can parse files containing multiple outputs from polyDFE, which enables an easier analysis. For this, multiple output files can be merged, for example, as follows:

The file run_polyDFE.txt contains the necessary polyDFE commands to obtain all of the above output files, which contain results for model A where:
-
central_chimp_A_nor_noeps: ri parameters are fixed to 1 and εan is fixed to 0;
-
central_chimp_A_nor: ri parameters are fixed to 1, but εan is estimated;
-
central_chimp_A_noeps: ri parameters are estimated, but εan is fixed to 0;
-
central_chimp_A: both ri and εan are estimated.
Corresponding files, as given in run_polyDFE.txt, can be obtained for model C and for a deleterious DFE model. Restricting either model A or model C to only deleterious mutations yields the same type of DFE: a reflected Γ distribution.
The output file central_chimp_A.txt from above can be parsed using the parseOutputR function (line 2) into a list where each entry corresponds to a different run of polyDFE found in the parsed file (line 3):

One entry in the list contains:
-
the name of the input file polyDFE was run on (line 5),
-
the DFE model used (line 6),
-
the best likelihood found and corresponding gradient (line 7),
-
the values of all parameters, including those that were not estimated (line 8),
-
which parameters have been estimated (line 9),
-
expectations for SFS and divergence (line 10),
-
estimates for α (line 11).
We can also get an overview of the gradients obtained during the optimization process:

The function getModelName (line 4) returns a string briefly describing the estimated model: which DFE model was used and whether the ri and εan parameters were estimated (+) or not (-). The runs using model A where εan was not estimated (A + r - eps), and models A and C where all parameters have been estimated (A + r + eps, C + r + eps), have gradients that are larger than 0.001. For these, running additional iterations of basin-hopping might lead to an improved solution (see Subheading 3.2).
4.3 Summarizing the DFE Estimated by polyDFE
The estimated DFEs under the different models can be discretized using the getDiscretizedDFER function (lines 3–5), and then plotted for visual comparison (lines 6–13). This is exemplified below, where the DFE is binned in six classes of 4Nes values, as shown in Fig. 1:

Estimated discretized DFEs for models A, A del (which contains only deleterious mutations) and C, where the ri parameters were estimated (+ r) or not (− r) and εan was estimated (+ eps) or not (− eps). The plot to the right has the y-axis on log scale

The majority of the mutations are typically deleterious and only few mutations have beneficial effects (Fig. 1). To more easily compare visually the discretized DFE, it can be useful to use a log-scale on the y-axis for plotting (lines 10–13, Fig. 1).
To obtain a discretized DFE, the continuous DFE has to be integrated. This is prone to numerical issues and sometimes the resulting discretized DFE does not sum exactly to 1. When this is the case, getDiscretizedDFE issues a warning.
4.4 Estimating α
polyDFE automatically calculates and outputs α. If divergence data was used when polyDFE was run, then both αdiv and αdfe are estimated, otherwise only the latter is calculated:

When a strictly deleterious DFE model is estimated, as by construction the DFE does not contain any beneficial mutations, the estimated αdfe is 0:

α can also be calculated in R from the estimated DFE using the function estimateAlpha (lines 5, 7). By default, αdfe is calculated (line 5), but divergence data can be parsed in R using the function parseDivergenceData (line 3) and provided to estimateAlpha (line 7), which then returns αdiv. We can compare the values that polyDFE outputted with the estimates obtained in R (lines 4–13). As polyDFE is implemented in C, the values for α returned by polyDFE are typically slightly different than the ones calculated in R (lines 8–13), though the difference should be minor:

Divergence data can contain misattributed polymorphism: SNPs that were misidentified as substitutions as they were fixed within the sample [1]. When calculating αdiv, polyDFE automatically corrects for this. Using R, this correction can be turned off (line 4), but typically the difference in the estimated αdiv is not very large (lines 3–7):

When calculating α, the expected number of substitutions that are either non-beneficial (deleterious or neutral) or beneficial is calculated. polyDFE and the R function estimateAlpha assume that a mutation that has a positive selection coefficient is beneficial. However, one could argue that a mutation with a very low positive selection coefficient is effectively neutral and only mutations that have a selection coefficient above a threshold Ssup should be considered as beneficial when calculating α [1, 12]. To obtain such estimates of α, the supLimit can be changed in R (lines 2–4). Setting a higher supLimit will mechanically decrease α, as fewer substitutions will be considered beneficial (lines 2–10):

5 Hypothesis Testing and Model Averaging
The estimation of DFE and α entails substantial statistical uncertainty. We discuss how to obtain the sampling variance of parameter estimates and approximate confidence intervals using a bootstrap approach. We also outline how to perform hypothesis testing and how to use model averaging as an alternative to the hard thresholding inherent to hypothesis testing. The flexibility of polyDFE allows performing model averaging with the advantage of generating parameter estimates where model uncertainty is also incorporated.
5.1 Bootstrap-Based Confidence Intervals
In principle, likelihood profiles can be obtained for one or more parameters by fixing these parameters to a set of values and maximizing the likelihood for all other parameters. Using polyDFE, this can be achieved by using the command line argument -i (see Subheading 3.1). The profile likelihood can then be used to obtain approximate confidence intervals for the parameters of interest.
In practice, we recommend to use a bootstrap approach:
-
1.
Generate 100–500 bootstrap datasets.
-
2.
Run polyDFE on these datasets.
-
3.
Calculate the sampling distribution of the ML estimates returned by polyDFE.
Although a crude likelihood profile can be obtained for a single parameter with as few as 20–30 runs of polyDFE, the bootstrap approach has the advantage of yielding sampling distributions for all parameters of interest in one go, as well as capturing the patterns of covariation between parameters.
Bootstraps are generated by re-sampling the data at the site level. More specifically, bootstrap datasets are obtained by parametric bootstrapping and assuming that all counts in the SFS and, possibly, divergence data are independent variables following a Poisson distribution, with means given by the observed data. This is in line with the modelling assumption that the number of mutations in each SFS entry and, possibly, divergence data follows a Poisson process. Using R, 500 bootstrap datasets can, for instance, be obtained using the commands:

which generate 500 datasets each stored in files cental_chimp_sfs_j (line 1) and bootstrap_central_j (line 2), with 1 ≤ j ≤ 500, respectively. The name of the output files can be optionally specified through outputfile (line 2).
To speed up the analysis when running polyDFE on bootstrap data, the command line argument -i (see Subheading 3.1) can be used to initialize the parameters to the values that were found when running polyDFE on the full dataset. Using the R function createInitLines (see the polyDFE manual for details), these values from a polyDFE output file can be written automatically to an initialization file that is then given to -i.
Once polyDFE is run on bootstrap data, confidence intervals for parameters, expected SFS, and discretized DFE can be obtained using the quantiles of the bootstrap distributions of the quantities of interest [14].
5.2 Hypothesis Testing
polyDFE returns ML estimates and therefore, likelihood ratio tests (LRT) and the Akaike information criteria (AIC) can be used to compare models.
The likelihood ratio test entails fitting two nested models using polyDFE, where one reduced model is a special case of a more general model. A p-value can be obtained by assuming that the log of the ratio of the maximum likelihoods of the two models follows a χ2 distribution parameterized by the difference in the number of degrees of freedom (i.e., number of estimated parameters) between the two models. A small p-value means that the reduced model is rejected in favor of the more parameter-rich model.
For instance, one can formally test for the occurrence of beneficial mutations by fitting two models A that differ by the maximum allowed scaled selection coefficient Smax, where in the general model Smax is freely estimated, while in the reduced model Smax is fixed to 0 and thus a deleterious DFE is estimated. Similarly, this can be done under model C by fixing the amount of beneficial mutations pb to 0. Note that the two reduced models under models A and C yield the same type of deleterious DFE, therefore testing for the occurrence of beneficial mutations under model C can be obtained by comparing the reduced model A with the general model C.
Using LRT requires that models are nested, which does not allow to test whether the full DFE model A or C fits the data better. For this, the Akaike information criteria can be used
where m is the number of estimated parameters (or degrees of freedom) and L is the maximum likelihood. Then the preferred model is the one with the minimum AIC value.
To test for the occurrence of beneficial mutations under model A, the p-values from the LRTs and AIC values can be obtained using the R function compareModels as follows:

The function sequentially compares model number j found in central_chimp_A.txt with model number j found in central_chimp_Del.txt (in the order of appearance in the files), which vary in the estimation of ri and εan, as detailed in Subheading 4.2 and run_polyDFE.txt.
The models found within central_chimp_A.txt and the other output files are also nested. For example, for models number 3 and 4, the ri parameters were estimated, but εan was either estimated (model number 4) or fixed to 0 (model number 3). The LRT for these two models can be obtained as follows:

The compareModels function automatically detects nestedness when the same DFE model was used. Recall that the deleterious DFEs obtained from either model A or model C are equivalent, but as the deleterious model was obtained from model A, to test for the occurrence of beneficial mutations under model C, nestedness has to be enforced by setting nested = TRUE:

As noted in Subheading 3.1, sometimes the ri parameters can also account for polarization errors. This is the case here, as we can observe that, when the ri parameters are not estimated and fixed to 1 (- r), the LRT and AIC indicate that inferring εan (+ eps) leads to a better fit of the data. However, when the ri parameters are estimated (+ r), εan is not needed for fitting the data (- eps). This is also supported by the estimated value of εan, which is very small when the ri parameters are estimated (+ r), but much larger otherwise (Table 1):

5.3 Model Averaging with polyDFE
Using model averaging provides a way to obtain the most honest estimates that account for model uncertainty. In particular, we can have a series of estimates that can differ by the DFE model assumed (A, B, C or D), whether or not distortions of the SFS are accounted for and possibly whether errors happen when polarizing the SNPs. One can choose the model most supported by the data using LRT or AIC as described in Subheading 5.2, but model averaging has the advantage of avoiding a strict thresholding where we decide that a given model is the best and exclude all other competing models. This might be necessary, for example, when the data contain only limited information about the DFE [15, 16] and therefore different models cannot be differentiated, due to AIC values that are very close (see also Subheading 5.4).
In practice, the parameter of interest x (such as any DFE parameter, α, entries in the discretized DFE or any other quantities) is estimated as xj under each model j that has an AIC value of AICj. These values are combined to yield a model-averaged estimate where the contribution of each model is averaged using Akaike weights [17]:
where ΔAICj is obtained as
When doing model averaging, models that are most supported by the data have a ΔAICj that is close to 0 and consequently a weight that is close to 1, while models fitting badly have high ΔAICj values and, accordingly, weights that shrink towards 0.
The AIC weights can be calculated using the R function getAICweights (line 3). Then the average value of, for example, αdiv (lines 4, 5) and αdfe (lines 6, 7) can be calculated as follows:

Note that getAICweights returns weights that are already rescaled so that they sum to 1, so the average value of α does not have to be normalized by the sum of the weights as in Eq. 1.
5.4 Note on Divergence Data
One of the advantages of polyDFE is that it does not require the use of divergence data for inferring the DFE and α. This is free of the assumption that the ingroup and outgroup share the DFE, which is needed when divergence data is used. Violating this assumption can lead to biases in the estimates [1]. However, divergence data is always available, as it is needed to orient the SNPs to calculate the derived SFS (see Subheading 2.1). So then the question arises on whether divergence data should be used or not in the inference.
The impact of using divergence data can be observed when investigating the AIC values (Table 2). When divergence data was not used, they are much closer to each other: the best 6 models are within an AIC difference of approximately 4, while when divergence data is used, only the first two models have an AIC difference of approximately 2, while the rest of the models have a much poorer fit. This is because using less data for the inference makes it more difficult to differentiate between the models. Using or not the divergence data also has a big impact on the estimates of α (Table 2):

The model-averaged αdfe (line 14) is estimated to be 0.388 when divergence data is not used, but only 0.225 when divergence data is used.
Deciding on whether or not divergence data should be used is not necessarily straightforward. The previous approaches of LRT and AIC are not applicable here, as we want to compare two models that are fitted on different datasets. One way is to compare the observed and expected SFS obtained under the estimated models. The best models found in both cases contain estimated ri parameters, but an εan that was fixed to 0. When divergence data was used, the best model is model C, while when divergence data is not used, the model of choice is model A (Table 2).
The observed SFS (line 1) is given as counts for the total length of the region, while the expected one (lines 2–4) is given per site, so before the comparison, the expected SFS has to be normalized by the length of the region (lines 5, 6). Using a log scale on the y-axis (lines 10, 11) and coloring the background in alternating colors (lines 12–16) makes it easier to visually compare the SFS for 1 ≤ i < n, where n is the sample size (line 7). Then the expected SFS counts scan be plotted (lines 19, 20) next to the observed SFS counts (line 21):

Visualizing the SFS (Fig. 2) can give insights into how well the models fit the data, but it does not give any statistical measure on how well the expected SFS matches the observed. To test that, we can use a χ2 goodness-of-fit test (lines 4–6). This indicates that both using the divergence data or not gives a good fit to the SFS, and that, in general, the selected SFS is more difficult to fit well (lines 8–10). The χ2 statistic could be used as a way to decide if divergence data should be used or not (lines 11–13) which shows that, overall, not using divergence data seems to give a closer fit to the data:

Observed and expected SFS for best fitted models when divergence data was used or not. The y-axis is on log scale

The above check can also be done using a model-averaged expected SFS, as detailed in analysis.R. Doing so does not change the above conclusion.
6 Conclusion
polyDFE provides a flexible likelihood framework to infer the DFE and properties of beneficial mutations from SFS and possibly divergence data. The estimation procedure accounts for uncertainty of the models and data imperfection. polyDFE is continually developed further with updates posted on https://github.com/paula-tataru/polyDFE and is currently being extended to test for heterogeneous DFE among species or gene categories within a single species.
Change history
27 February 2021
Chapter 2, “Processing and Analyzing Multiple Genomes Alignments with MafFilter,” was previously published without including the Electronic Supplementary Material. This has now been included in the revised version of this book.
References
Tataru P, Mollion M, Glémin S, Bataillon T (2017) Inference of distribution of fitness effects and proportion of adaptive substitutions from polymorphism data. Genetics 207(3):1103–1119
Bataillon T, Bailey SF (2014) Effects of new mutations on fitness: insights from models and data. Ann New York Acad Sci 1320(1):76–92
Gossmann TI, Song BH, Windsor AJ, Mitchell-Olds T, Dixon CJ, Kapralov MV, Filatov DA, Eyre-Walker A (2010) Genome wide analyses reveal little evidence for adaptive evolution in many plant species. Mol Biol Evol 27:1822–1832
Castellano D, Coronado-Zamora M, Campos JL, Barbadilla A, Eyre-Walker A (2016) Adaptive evolution is substantially impeded by Hill-Robertson interference in Drosophila. Mol Biol Evol 33:442–455
Hernandez RD, Williamson SH, Bustamante CD (2007) Context dependence, ancestral misidentification, and spurious signatures of natural selection. Mol Biol Evol 24(8):1792–1800
Keightley PD, Campos JL, Booker TR, Charlesworth B (2016) Inferring the frequency spectrum of derived variants to quantify adaptive molecular evolution in protein-coding genes of Drosophila melanogaster. Genetics 203(2):975–984
Keightley PD, Jackson BC (2018) Inferring the probability of the derived versus the ancestral allelic state at a polymorphic site. Genetics 209(3):897–906
Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, Hubisz MJ, Fledel-Alon A, Tanenbaum DM, Civello D, White TJ, Sninsky JJ, Adams MD, Cargill M (2005) A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol 3(6):e170
James JE, Piganeau G, Eyre-Walker A (2016) The rate of adaptive evolution in animal mitochondria. Mol Ecol 25(1):67–78
Bataillon T, Duan J, Hvilsom C, Jin X, Li Y, Skov L, Glemin S, Munch K, Jiang T, Qian Y, Hobolth A (2015) Inference of purifying and positive selection in three subspecies of chimpanzees (Pan troglodytes) from exome sequencing. Genome Biol Evol 7(4):1122–1132
Bierne N, Eyre-Walker A (2003) The problem of counting sites in the estimation of the synonymous and nonsynonymous substitution rates: implications for the correlation between the synonymous substitution rate and codon usage bias. Genetics 165(3):1587–1597
Galtier N (2016) Adaptive protein evolution in animals and the effective population size hypothesis. PLoS Genet 12(1):e1005774
Wales DJ, Doye JP (1997) Global optimization by basin-hopping and the lowest energy structures of Lennard-Jones clusters containing up to 110 atoms. J Phys Chem A 101(28):5111–5116
Efron B, Tibshirani RJ (1993) An introduction to the bootstrap: monographs on statistics and applied probability, vol 57. Chapman and Hall/CRC, New York/London
Boyko AR, Williamson SH, Indap AR, Degenhardt JD, Hernandez RD, Lohmueller KE, Adams MD, Schmidt S, Sninsky JJ, Sunyaev SR, White TJ (2008) Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet 4(5):e1000083
Wilson DJ, Hernandez RD, Andolfatto P, Przeworski M (2011) A population genetics-phylogenetics approach to inferring natural selection in coding sequences. PLoS Genet 7(12):e1002395
Posada D, Buckley TR (2004) Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst Biol 53(5):793–808
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
1 Electronic supplementary material
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this protocol

Cite this protocol
Tataru, P., Bataillon, T. (2020). polyDFE: Inferring the Distribution of Fitness Effects and Properties of Beneficial Mutations from Polymorphism Data. In: Dutheil, J.Y. (eds) Statistical Population Genomics. Methods in Molecular Biology, vol 2090. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-0199-0_6
Download citation
DOI: https://doi.org/10.1007/978-1-0716-0199-0_6
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-0198-3
Online ISBN: 978-1-0716-0199-0
eBook Packages: Springer Protocols