
Abstract
Systemic chemotherapy is the only current method of treatment that provides some chance for long-term survival in patients with advanced or metastatic cancer. γ-Tocotrienol is a natural form of vitamin E found in high concentrations in palm oil and displays potent anticancer effects, but limited absorption and transport of by the body has made it difficult to obtain and sustain therapeutic levels in the blood and target tissues. Statins are inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMGCoA) reductase and are an example of a promising cancer chemotherapeutic agent whose clinical usefulness has been limited due to high-dose toxicity. Similarly, erlotinib and gefitinib are anticancer agents that inhibit the activation of individual HER/ErbB receptor subtypes, but have shown limited clinical success because of heterodimerization between different EGF receptor family members that can rescue cancer cells from agents directed against a single receptor subtype. Recent studies have investigated the anticancer effectiveness of low-dose treatment of various statins or EGF receptor inhibitors alone and in combination with γ-tocotrienol on highly malignant +SA mouse mammary epithelial cells in vitro. Combined treatment with subeffective doses of γ-tocotrienol with these other chemotherapeutic agents resulted in a synergistic inhibition of +SA cell growth and viability. These findings strongly suggest that combined treatment of γ-tocotrienol with other anticancer agents may not only provide an enhanced therapeutic response but also provide a means to avoid the toxicity, low bioavailability, or limited therapeutic action associated with high-dose monotherapy.
Similar content being viewed by others


Introduction
Cancer is defined as unregulated cell proliferation that leads to a disruption in normal function and viability of surrounding tissues and organs and ultimately leads to death. Initial stages of cancer are confined to the tissue site of origin, whereas advanced cancer invades surrounding tissues and can be distributed through the circulatory and lymphatic systems to colonized distant tissues and form metastasis. Collectively, cancer is second only to cardiovascular disease as the most prevalent cause of death in the United States (American Cancer Society 2009). The development of cancer results from mutations in the cellular genome that can occur through random endogenous mechanisms or exposure to environmental influences. Breast cancer is the most prevalent malignancy in women in the world (Marshall 1993), and although mutations in specific tumor repressor genes (BRCA1 and BRCA2) predispose women to breast cancer, these genetic mutations are quite rare and account for only 10–15% of all breast cancers (Vogel 1993). The cause for the other 85–90% breast cancers in women for the most part remains unknown. Cancer is a multistage process that is initiated by a single genomic mutation, followed by subsequent mutations that lead to a progression in malignancy, characterized by increased anaplastic histological morphology, resistance to drug or endocrine therapy, and enhanced metastatic potential (Medina 1974; Nagasawa et al. 1976; Russo and Russo 1978; Sylvester et al. 1983, 1986a; Nandi et al. 1995).
At present, anticancer therapy has focused on developing drugs directed against specific molecular targets responsible for neoplastic transformation and progression. Much evidence has accumulated over the past few years, which demonstrates dietary supplementation with specific forms of vitamin E, particularly tocotrienols, can provide significant protection at multiple stages of mammary carcinogenesis (Rogers and Lee 1986; Sylvester et al. 1986b; Sundram et al. 1990; Cottrell 1991; Nakayama et al. 1993; Goh et al. 1994). These studies strongly suggest that tocotrienols may have great potential in the prevention and treatment of cancer because they possess multiple anticancer mechanisms of action directed against all stages of tumor initiation and progression. It is also well understood and appreciated that anticancer chemotherapy is most effective when multiple drugs with complimentary mechanisms of action are given in combination. Combination chemotherapy optimizes the effectiveness of each drug to bring about a complimentary and synergistic therapeutic response, while at the same time, reducing toxic adverse side effects associated with high-dose monotherapy. Various investigations have shown that combined low-dose tocotrienol treatment with specific chemotherapeutic agents displays significantly enhanced anticancer effects, as compared to that observed from individual treatments alone (Bachawal et al. 2010a, b; Shirode AB and Sylvester 2010; Wali and Sylvester 2007; Wali et al. 2009a, b).
Statins and cancer
Stains constitute a class of drugs widely used in clinics for the treatment of hypercholesterolemia. Statins such as simvastatin, lovastatin, mevastatin, and pravastatin are naturally produced by certain fungi, whereas cerivastatin, atorvastatin, fluvastatin, rosuvastatin, and pitvastatin are derived synthetically. Irrespective of their source, statins are known to decrease high blood cholesterol levels by suppressing the biosynthesis of cholesterol in the liver. Statins share structural resemblance with 3-hydroxy-3-methylglutaryl-coenzyme A (HMGCoA) and competitively inhibit HMGCoA reductase, the rate limiting enzyme in mevalonate pathway for cholesterol synthesis (Goldstein and Brown 1990). Interestingly, the mevalonate pathway is constitutively active in malignant cells due to elevated and unregulated HMGCoA reductase activity in various types of cancer (Mo and Elson 2004). In addition to cholesterol, the mevalonate pathway produces abundant non-sterol products that are required for isoprenylation of key signaling proteins such as Ras, Rap, and Rab. Isoprenylation results in anchoring these proteins to the interior of the plasma membrane, which is required for their activation and initiation of downstream mitogenic signaling pathways vital for cellular growth and survival. Furthermore, a higher portion of reductase activity in many kinds of cancer cells is directed toward the synthesis of nonsterol intermediates that are required for malignant cell growth. Statins act to block the mevalonate pathway by inhibiting HMGCoA reductase and subsequent isoprenylation of Ras family proteins (Graaf et al. 2004). For this reason, the anticancer potential of statins has been extensively studied not only because of their actions to inhibit HMGCoA reductase but also because mutate from of Ras proteins have been found to be present in at least 30% of human cancers.
Statins display antiproliferative and apoptotic activity against many types of cancer cell lines (Graaf et al. 2004; Campbell et al. 2006; Maltese et al. 1985; Seeger et al. 2003). In addition to inhibiting prenylation of Ras family proteins, statins have also been found to inhibit the EGF receptor function in head and neck squamous cell carcinomas and breast adenocarcinoma cells (Mantha et al. 2005). Statins have been shown to suppress downstream signaling of EGF receptors and attenuate ERK1/2, JNK, PI3K/Akt, and p38 activation in breast and other types of cancer (Campbell et al. 2006). Statin inhibition of these mitogenic pathways is also associated with a reduction in cyclin D1, CDL2, and CDK4 and increase in p21 and p27, hypophosphorylation in Rb, and cell cycle arrest in G1 in breast, colon, prostate, head and neck, and glioblastoma tumor cells (Mo and Elson 2004). At higher doses, statins initiate apoptosis by suppressing Bcl-2 and Bcl-xL and increase Bax and Bad expression, as well as caspase-3 activation, and combined treatment with mevalonate was found to reverse these effects (Mo and Elson 2004).
The antitumor efficacy of statins has also been shown in various in vivo models (Shibata et al. 2003, 2004). Statins inhibit the growth of human myeloid leukemia cells in severe combined immunodeficient mice, syngeneic mouse lung cancer, glioma cells in nude mice, and ErbB2-transformed mouse mammary cancer (Shibata et al. 2003, 2004). Preclinical studies in animals including mouse, rat, rabbit, and dog revealed that circulating concentrations up to 4 μM lovastatin were associated with anorexia and death (Graaf et al. 2004; Gerson et al. 1989; Kornbrust et al. 1989). Clinical trials with prostate, breast, colorectal, ovarian, and lung cancer patients determined that high-dose treatment was associated with severe myopathy with the maximum dose of lovastatin that could be tolerated to be 25 mg/kg, which produced a peak plasma concentration of 3.9 μM (Thibault et al. 1996). Since the dose of lovastatin required to inhibit growth and induce apoptosis of cancer cells in culture ranged between 0.1 and 100 μM, toxic doses of lovastatin must be used to effectively treat cancer patients (Thibault et al. 1996; Chan et al. 2003). Taken together, these findings indicate that statin monotherapy is too toxic to be used as monotherapy in the treatment of cancer.
Combined treatment of statins with γ-tocotrienol against mammary tumor cells
It is well established that statins are potent inhibitors of HMGCoA reductase, an enzyme catalyzing the conversion of HMGCoA to mevalonate, the rate limiting step in cholesterol synthesis. However, γ-tocotrienol has also been shown to induce the posttranscriptional down-regulation of HMGCoA reductase, resulting in a significant decrease in cholesterol synthesis in a similar manner as 25-hydroxycholesterol (Parker et al. 1993). Specifically, tocotrienols appear to reduce HMGCoA reductase activity by triggering both the retention of sterol regulatory element binding proteins (SREBP) on the endoplasmic reticulum and the degradation of HMGCoA reductase, thereby inducing the reduction in both mRNA and protein HMGCoA reductase levels (Goldstein et al. 2006). In contrast, statins are competitive inhibitors of HMGCoA reductase and act to directly inhibit HMGCoA reductase activity. This effect of statins causes a reduction in cholesterol, the negative feedback and subsequently induces SREBP cleavage and inhibits HMGCoA reductase degradation, resulting in an increase in both mRNA and protein HMGCoA reductase expression (Goldstein et al. 2006) (Fig. 1). Since statins and γ-tocotrienol suppress HMGCoA reductase activity through independent mechanisms, it was logical to conclude that combined low-dose treatment with these agents may produce an additive or even synergistic anticancer effects, while at the same time avoiding high-dose side effects. Initial studies were conducted to examine the growth inhibitory effects of low-dose treatment with various statins alone and in combination with γ-tocotrienol against the highly malignant mouse +SA mammary tumor cells in culture maintained in serum-free defined media using EGF as a mitogen (Wali and Sylvester 2007). Studies were also conducted to determine the intracellular signaling mechanisms that were involved in mediating the inhibitory effects of combined low-dose statin and γ-tocotrienol treatment on EGF-dependent proliferation and survival (Wali and Sylvester 2007).

The role of γ-tocotrienol and statins in attenuating 3-hydroxy-3methyl-glutaryl-coenzyme A (HMGCoA) reductase activity and downstream progression of the mevalonate synthesis pathway
Results from these studies showed that treatment with 2–8 μM simvastatin, lovastatin, or mevastatin alone significantly inhibited, whereas treatment with 10–100 μM pravastatin alone had no effect on EGF-dependent +SA mammary tumor cell growth (Wali and Sylvester 2007). In addition, treatment with growth inhibitory doses (2–8 μM) of simvastatin, lovastatin, or mevastatin was not found to be cytotoxic to +SA mammary tumor cells and only reduce viable +SA cell number when cells were exposed to extremely high, clinically irrelevant doses (100–200 μM). Although treatment with 2–8 μM simvastatin, lovastatin, or mevastatin alone was found to significantly inhibit +SA mammary tumor cell growth, an oral dose of 25 mg/kg or higher is required to achieve the same levels of these statins in the blood of humans, and treatment with doses of 25 mg/kg or higher in humans is associated with severe adverse side effects, particularly myotoxicity (Thibault et al. 1996). Figure 1 illustrates the role of γ-tocotrienol and statins in attenuating 3-hydroxy-3methyl-glutaryl-coenzyme A (HMGCoA) reductase activity and downstream progression of the mevalonate synthesis pathway.
Subsequent studies showed that combined treatment with a subeffective dose (0.25 μM) of simvastatin, lovastatin, or mevastatin with subeffective doses (0.25–2.0 μM) of γ-tocotrienol resulted in a synergistic inhibition of +SA cell growth (Wali and Sylvester 2007). Interestingly, it was observed that treatment with much higher doses (10–100 μM) of pravastatin alone had no growth inhibitory effects against +SA cells, but combined treatment of 10 μM pravastatin with subeffective doses of γ-tocotrienol resulted in a significant dose-responsive inhibition of +SA cell proliferation (Wali and Sylvester 2007). These findings again demonstrate that pravastatin is far less potent anticancer agent either alone or in combination with γ-tocotrienol, as compared to the other statins tested. Furthermore, combined low-dose treatment of γ-tocotrienol and individual statins were not found to be cytotoxic. These findings may have very important clinical implications in that combined low-dose statin and γ-tocotrienol treatment may not only provide synergistic anticancer activity in the treatment of breast cancer but also may be devoid of the adverse side effects and severe myotoxicity that is associated with high-dose statin use.
In addition, results showed that treatment with subeffective doses of γ-tocotrienol or various statins alone had no effect on EGF-dependent mitogenic signaling in +SA mammary tumor cells (Fig. 2). However, when these subeffective doses of γ-tocotrienol and simvastatin, lovastatin, or mevastatin were used in combination, a marked suppression in the relative intracellular levels of phosphorylated (activated) p44 MAPK, p54/46 JNK, p38, and Akt was observed (Wali and Sylvester 2007), whereas combined treatment of γ-tocotrienol and pravastatin resulted only in a large suppression of phosphorylated p38 and had no significant effect on phosphorylated (activated) p44 MAPK, p54/46 JNK, and Akt expression in these cells (Fig. 2). These results may partly provide the explanation why pravastatin displays less potent anticancer activity than other statins when given alone or in combination with γ-tocotrienol (Wali and Sylvester 2007). In conclusion, results for these studies demonstrate the synergistic antiproliferative effects of combined low-dose treatment of γ-tocotrienol and individual statins against mammary tumor cells. Furthermore, these findings strongly suggest that combined γ-tocotrienol and statin therapy may provide significant health benefits in the prevention and/or treatment of breast cancer in women, while at the same time avoiding myotoxicity that is associated with high-dose statin treatment.

From Wali and Sylvester (2007) with permission. Effects of range of subeffective doses of γ-tocotrienol when given alone or in combination with a subeffective dose of individual statins on the growth of neoplastic +SA mammary epithelial cell growth. Cells were initially plated at a density of 5 × 104 cells/well (6 wells/group) in 24-well culture plates and exposed to treatments for a 4-day period. Afterward, viable cell number was determined using MTT colorimetric assay. Vertical bars indicate the mean cell count ±SEM in each treatment group. *P < 0.05, as compared with vehicle-treated controls
Combined treatment of statins and γ-tocotrienol induces cell cycle arrest in mammary tumor cells
Cell cycle progression involves a series of highly regulated phases (G1, S, G2, and M phases), which results in DNA replication and cell division (Baserga and Wiebel 1969; Norbury and Nurse 1992). The transition between these phases is controlled by cyclin-dependent kinases (CDKs) and their regulatory binding proteins called cyclins (Massague 2004). Cyclin/CDK complexes regulate cell cycle entry from G1 to S phase and phosphorylate and inactivate Rb, the tumor suppressor protein that suppresses cell cycle progression (Massague 2004). The cyclin/CDK/Rb regulatory axis is dysfunctional in nearly all forms of human cancer (Shapiro 2006). The primary mitogen-dependent signaling pathway involved in regulating the expression of cyclin D1 is the Akt and MAPK cascades. Since combined low-dose statin and γ-tocotrienol treatment was previously shown to suppress the activation of Akt and MAPK signaling in +SA mammary tumor cells (Wali and Sylvester 2007), studies were conducted to determine whether these effects were associated with a reduction in cyclin D1 levels and subsequent induction of cell cycle arrest in G1 in +SA mammary tumor cells (Wali et al. 2009b). Additional studies were also conducted to determine the effects of combined statin and γ-tocotrienol treatment on intracellular levels of CDK2, CDK4, CDK6, p15, p21, p27, and hyperphosphorylated Rb protein (Wali et al. 2009b).
Experimental findings showed that combined treatment with low doses of statins and γ-tocotrienol induces cell cycle arrest in G1 in highly malignant +SA mammary epithelial cells in culture (Wali et al. 2009b). Cell cycle analysis by flow cytometry showed that combined γ-tocotrienol and statin treatment induced cell cycle arrest without inducing apoptosis (Wali et al. 2009b). These findings are in agreement with previous studies that showed that similar treatments with statins and γ-tocotrienol were found to be cytostatic but not cytotoxic to these cells (Wali and Sylvester 2007) and further support the suggestion that unwanted toxicity associated with high-dose statin monotherapy might be avoided by using low-dose statin therapy in combination with another anticancer agent such as γ-tocotrienol.
Additional studies showed that these cytostatic effects were associated with a large decrease in the relative levels of cyclin D1, CDK2, and hyperphosphorylated Rb protein and a corresponding increase in p27 levels (Wali et al. 2009b). Since similar effects were observed when γ-tocotrienol was combined with simvastatin, lovastatin, mevastatin, or pravastatin, these effects appear to be common to all statins and not a unique characteristic displayed by an individual statin family member. Cell cycle transition from G1 to S phase is primarily regulated by cyclin D1 in complex with CDKs. Mitogenic stimuli increase the intracellular levels of cyclin D1, which then form an assembly with CDK4 or CDK6, and ultimately leading to the hyperphosphorylation of Rb protein (Sherr 1993; Matsushime et al. 1994). Elevations in cyclin D1 result in a corresponding increase in cyclin D1/CDK4 activity and hyperphosphorylation of Rb protein, whereas blockade of cyclin D1 activation blocks the hyperphosphorylation of Rb protein and results in cell cycle arrest in G1 (Grillo et al. 2006). Overexpression of cyclin D1 has been observed in approximately 50% of all breast cancers (Hall and Peters 1996; Arnold and Papanikolaou 2005; Caldon et al. 2006), whereas suppression of cyclin D1 activity significantly inhibits mammary tumorigenesis (Bartkova et al. 1994; Arber et al. 1997). Combined statin and tocotrienol treatment caused a relatively large reduction in cyclin D1 expression in malignant +SA mammary epithelial cells (Wali et al. 2009b). Active cyclin D1-CDK4/6 and cyclin E-CDK2 complexes sequentially phosphorylate and inactivate the Rb protein to initiate E2F-dependent transcription, a requirement for progression into S phase, and combined γ-tocotrienol and statin treatment resulted in a marked reduction in CDK2 levels and a reduction in Rb phosphorylation in +SA mammary tumor cells (Wali et al. 2009b).
EGF stimulates Akt and MAPK mitogenic signaling in +SA cells (Shah and Sylvester 2005), and activation of these pathways stimulates cell cycle progression from G1 to S phase by stimulating cyclin D1 activity and subsequent hyperphosphorylation of Rb protein (Massague 2004; Torii et al. 2006). Activation of Akt increases cyclin D1 transcriptional and protein expression (Liang and Slingerland 2003), and increase glycogen synthase kinase 3β (GSK3β) phosphorylation, and thereby preventing GSK3β from phosphorylating and destabilizing the cyclin D1 protein (Massague 2004). Akt also phosphorylates FOXO transcription factors that act to repress cyclin D1 expression and induce p27 expression (Tran et al. 2003), whereas MAPK activation phosphorylates and stabilizes c-myc, a transcription factor that induces cyclin D1 and decreases CKI expression (Sears and Nevins 2002; Coleman et al. 2004). In summary, these results demonstrated that γ-tocotrienol acts synergistically with statins to induce cell cycle arrest in G1 by significantly inhibiting Akt and MAPK mitogenic signaling, which causes a large decrease in intracellular levels of cyclin D1, CDK2 and hypophosphorylation of Rb and increased expression in p27 (Wali et al. 2009b).
Role of mevalonate synthesis in mediating the anticancer effects of combined statin and γ-tocotrienol treatment
Based on the finding in previous investigations, it is now clearly established that combined low-dose treatment of statins with γ-tocotrienol synergistically inhibited the growth of highly malignant +SA mammary epithelial cells in culture (Wali and Sylvester 2007; Wali et al. 2009b), and these effects result from a reduction in MAPK and Akt mitogenic signaling (Sylvester and Shah 2005; Sylvester and Theriault 2003). Furthermore, this combination therapy induced G1 cell cycle arrest by inducing a relatively large increase in p27 expression and corresponding decrease in cyclin D1 expression, as well as the hypophosphorylation of Rb (Wali et al. 2009b). Nevertheless, identification of the specific intracellular target responsible for mediating the antiproliferative effects of combined statins and γ-tocotrienol has not yet been identified. Since statins and γ-tocotrienol have both been shown to suppress HMGR activity, it was hypothesized that inhibition of mevalonate synthesis may be ultimately responsible for mediating the growth-inhibiting effects of combined therapy with these agents. Therefore, studies were conducted to characterize the role of mevalonate synthesis in mediating the antiproliferative effects of combined statin and γ-tocotrienol treatment in +SA mammary tumor cells.
Experimental results showed that the growth inhibitory effects of combined low-dose statin and γ-tocotrienol treatment on +SA mammary tumor cells are directly related to the suppression of HMGCoA reductase activity (Wali et al. 2009a). Specially, combined treatment with these agents resulted in a significant inhibition of +SA cell growth, and corresponding decrease in total HMGCoA reductase, Rap1A and Rab6 prenylation, and MAPK signaling, and supplementation with mevalonate reversed these effects (Wali et al. 2009a). However, these studies also showed that the growth inhibitory effects resulting from treatment with a moderated dose (4 μM) of γ-tocotrienol is not dependent on a reduction in HMGCoA reductase activity because this treatment had little or no effect on protein prenylation, and mevalonate supplementation did not reverse these antiproliferative effects (Wali et al. 2009a). These findings clearly demonstrate that the synergistic antiproliferative effects of combined low-dose statin and γ-tocotrienol treatment result directly from an inhibition of HMGCoA reductase activity and subsequent suppression of mevalonate synthesis (Fig. 3). The ability of subeffective doses of γ-tocotrienol to synergistically enhance the growth inhibitory effects of individual statins can be partly explained by the finding that statin inhibition of HMGCoA reductase activity is associated with triggering a compensatory feedback mechanism that results in an up-regulation of HMGCoA reductase expression (Brown et al. 1978; Nakanishi et al. 1988), and combined treatment with γ-tocotrienol blocks this self-limiting statin effect. In summary, the synergistic antiproliferative effects of combined low-dose statin and γ-tocotrienol treatment against +SA mammary tumor cells results from suppression of HMGCoA reductase activity and subsequent reduction in mevalonate synthesis (Fig. 3). The synergistic antiproliferative activity of these agents appears to be due to the cooperative action of γ-tocotrienol to cause a down-regulation of HMGCoA reductase levels and statins to directly inhibit HMGCoA reductase activity.

From Wali et al. (2009a) with permission. Effects of mevalonate supplementation on the growth of +SA mammary tumor cells. Cells were plated at a density of 5 × 104 cells/well (6 wells/group) in 24-well culture plates and exposed to control (C), 0.25 μM simvastatin (S), 0.25 μM lovastatin (L), 0.25 μM mevastatin (M), 10 μM pravastatin (P), 2 μM γ-tocotrienol (T), 1 μM mevalonate (m1), or 2 μM mevalonate (m2) alone or combination with individual statins (S, L, M or P) and/or γ-tocotrienol (T) for a 4-day culture period. Afterward, viable cell count was determined using the MTT assay. Vertical bars indicate the mean cell count ±SEM in each treatment group. *P < 0.05 as compared with respective combination statin and γ-tocotrienol treatment groups not exposed mevalonate supplementation
EGF receptors and cancer
The family of EGF receptors is composed of typical receptor tyrosine kinases that are classified as HERs in humans and ErbBs in rodents. The normal functions of EGF receptors include regulating cell proliferation, differentiation, migration, and apoptosis. The EGF family consists of four transmembrane receptors indentified as EGF receptor (HER1/ErbB1), HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4). All EGF receptors have a glycosylated extracellular ligand binding domain where ligands can bind and activate the receptor, a transmembrane domain embedded within the plasma membrane responsible for the cooperation between different EGF receptor family members and an intracellular catalytic tyrosine kinase domain that allows the interaction and phosphorylation of intracellular substrates involved with the regulation of signaling pathways (Normanno et al. 2003; Schlessinger 2000).
Ligands that bind to and activate EGF receptors are characterized by the presence of an EGF-like domain with three disulfide bonds that are required for binding and activating the receptor (Yarden 2001). These growth factors are formed by the proteolytic cleavage of the soluble transmembrane precursors that are biologically active and can activate the adjacent receptors through juxtacrine stimulation (Normanno et al. 2003; Massague and Pandiella 1993). A large number of ligands have been identified that can activate HER1/ErbB1, HER3/ErbB3, and HER4/ErbB4, yet no known ligand has been identified that can bind and activate HER2/ErbB2 directly. In contrast, HER/ErbB3 can bind to various likes like NRGs, but lack the intracellular tyrosine kinase domain. However, both HER2/ErbB2 and HER3/ErbB3 can participate in signal transduction by acting as co-receptors in the formation of heterodimers. Dimerization is an important step in the activation of EGF receptors, and the intensity of activation of downstream signaling pathways is dependent on the type of dimer formed. Ligand binding to the extracellular domain of EGF receptors results in the pairing and formation of receptor dimers. Dimers formed by the pairing of two of the same EGF receptors are call homodimers, while those formed by the pairing of different EGF receptors are call heterodimers. Even in the absence of ligand binding to HER2/ErbB2, this receptor exists in a favorable dimerization conformation and is a preferred dimerization partner for all other EGF receptors (Karunagaran et al. 1996; Tzahar et al. 1997). Although HER3/ErbB3 lacks intrinsic tyrosine kinase activity, ligand binding to this receptor will induce dimerization with other EGF receptors to initiate signal transduction.
In many types of cancer cells, abnormal activation or overexpression of EGF receptors is characterized by unregulated cell proliferation, enhanced survival, and metastasis. Various strategies have been developed to target EGF receptors including monoclonal antibodies (trastuzumab) directed toward the extracellular domain and tyrosine kinase inhibitors (erlotinib and gefitinib), which compete with ATP and block the ATP bind site on the receptor. However, clinical success of agents used to target EGF receptors has been disappointing because of the heterodimer cooperation existing between different EGF receptor family members and also other tyrosine kinase receptors, which can rescue the cancer cells from the antiproliferative effects of agents that target a single EGF receptor subtype (Olayioye et al. 2000). Therefore, a great deal of effort has been recently focused on understanding of EGF receptor heterodimer cooperation in order to improve the therapeutic effectiveness of these agents.
γ-Tocotrienol and EGF receptors in breast cancer
It is well established that treatment with relatively low doses of γ-tocotrienol significantly inhibited EGF-dependent neoplastic +SA mouse mammary epithelial cell growth, while having no effect on cell viability (Shah and Sylvester 2005), and that these antiproliferative effects of γ-tocotrienol resulted from a suppression of EGF-dependent activation of the PI3K/PDK-1/Akt mitogenic signaling pathway (Shah and Sylvester 2005). However, the exact intracellular sites of action targeted by γ-tocotrienol to inhibit PI3K/PDK1/Akt mitogenic signaling had not been determined. It was also possible that the antiproliferative effects of γ-tocotrienol occur upstream of the PI3K/PDK-1/Akt mitogenic signaling pathway at the level of the EGF receptor. Studies were conducted to determine the intracellular site of action mediating the inhibitory effects of γ-tocotrienol on EGF-dependent mammary tumor cell growth. Figure 4 illustrates the role of γ-tocotrienol, gefitinib, and erlotinib in suppressing EGF receptor-dependent mitogenic signaling pathways.

Role of γ-tocotrienol, gefitinib, and erlotinib in suppressing EGF receptor-dependent mitogenic signaling pathways
Results from these studies showed that the antiproliferative effects of γ-tocotrienol in +SA mammary tumor cells were mediated through a suppression of ErbB3 receptor tyrosine phosphorylation and subsequent reduction in PI3K/PDK-1/Akt mitogenic signaling (Samant and Sylvester 2006). Since elevated Akt signaling is associated with advanced tumor progression and poor prognosis in breast cancer patients (Downward 1998) and ErbB3 heterodimers are the most potent activators of the PI3K/PDK-1/Akt mitogenic pathway (Toker 2000), these findings strongly suggest γ-tocotrienol may have potential therapeutic value when used in combination with other chemotherapeutic agents directed against selected ErbB receptor activation in the treatment of breast cancer in women. This suggestion is further supported by in vitro and in vivo studies that have shown that combined therapy directed against multiple members of the ErbB receptor family display significantly greater antitumor activity than agents that target only a single ErbB receptor (Normanno et al. 2002; Britten 2004).
Combined treatment of γ-tocotrienol with EGF receptors inhibitors against mammary tumor cells
Since previous studies showed that the antiproliferative effects of γ-tocotrienol are associated with suppression of ErbB3 receptor activation and mitogenic signaling, studies were conducted targeting multiple ErbB receptors with combined treatment of γ-tocotrienol with ErbB receptor inhibitors to determine whether combined treatment provided a greater anticancer response as compared to monotherapy that targets only a single ErbB receptor subtype. Results from these studies showed that treatment with 0.5–3 μM of erlotinib, 1–3 μM gefitinib, or 3.5 μM γ-tocotrienol significantly inhibited EGF-dependent +SA cell growth (Bachawal et al. 2010a, b).Treatment with 0–250 μg/ml trastuzumab alone or in combination with 0–3 μM γ-tocotrienol had no effect on +SA cell growth or viability. The exact reason why trastuzumab was ineffective in suppressing +SA cell growth is presently unknown, but may be related to the fact that not all the breast cancer cells are sensitive to the trastuzumab treatment. Previous investigations have shown that trastuzumab is effective only in cancer cells that are primarily dependent on oncogenic HER/ErbB2 receptor mitogenic signaling (Hermes et al. 2008), whereas other studies using the NIH3T3-HER2 mouse model reported that cancer cells can be ErbB2 dependent, yet still display resistance to trastuzumab treatment (Hermes et al. 2008).
Treatment with 3 μM γ-tocotrienol, 0.25 μM erlotinib, or 0.5 μM gefitinib alone had no effect on +SA cell growth or viability, whereas combined treatment with these agents resulted in a synergistic inhibition of mammary tumor cell growth and initiated apoptosis (Bachawal et al. 2010b). Previous studies have shown that treatment with 10–20 μM γ-tocotrienol was required to initiate apoptosis in +SA cells (McIntyre et al. 2000a, b). Similarly, erlotinib and gefitinib have also been shown to induce apoptosis, but at much greater doses (≥1 μM) than those used in the present study (Ling et al. 2008; Okubo et al. 2004).
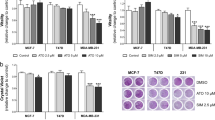
Subsequent studies were conducted to characterize ErbB receptor levels and activation in +SA mammary tumor cells. Results showed that ErbB1-4 receptors are all expressed in these cells and display tyrosine phosphorylation within 5–10 min following EGF exposure. ErbB1-4 expression gradually decreased between 30 and 60 min following EGF exposure due to the growth factor–induced receptor endocytosis and degradation (Bachawal et al. 2010b). Combined low-dose treatment with γ-tocotrienol and erlotinib or gefitinib caused a large suppression of ErbB3 and ErbB4 receptor phosphorylation and activation (Fig. 5). Overexpression of ErbB3 and ErbB4 receptors is associated with poor patient prognosis in various types of cancers (Carraway and Cantley 1994; Knowlden et al. 1998), and these findings indicate that combination therapy with agents that target multiple ErbB receptors effectively interferes with the heterodimer cooperation that exists between the various ErbB receptors (Fig. 5). Furthermore, since clinical use of agents that target only one member of the ErbB family of receptors have shown limited success in the treatment of cancer, these results strongly suggest that combined treatment of γ-tocotrienol with erlotinib or gefitinib targets multiple ErbB receptors and provides synergistic inhibition of mammary tumor cell growth and induces apoptosis. These findings strongly suggest that combination therapy may greatly improve therapeutic responsiveness in breast cancer patients.

From Bachawal et al. (2010a), with permission. Effects of combined γ-tocotrienol and erlotinib or gefitinib treatment on +SA cell growth (a, c) and ErbB receptor levels (b, d). Vertical bars indicate the viable cell count ±SEM in each treatment group. *P < 0.05 as compared to vehicle-treated control group (a, c). Western blot analysis measured total and phosphorylated levels of ErbB3 and ErbB4 receptors and α-tubulin levels (b, d). α-Tubulin was visualized to ensure equal sample loading in each lane. Each Western blot is a representative image of data obtained for experiments that were repeated at least three times
Combined γ-tocotrienol and EGF receptor inhibitor effects on EGF-dependent mitogenic signaling pathways
Studies were conducted to determine the specific intracellular signaling pathways involved in mediating the growth inhibitory effects of combined γ-tocotrienol and erlotinib or gefitinib treatment in +SA mammary tumor cells and the effects of this combination treatment on the EGF-dependent EGF receptor mitogenic signaling. Results showed that treatment with 3 μM γ-tocotrienol, 0.25 μM erlotinib, or 0.5 μM gefitinib alone did not alter the relative intracellular levels of phospho-Stat3, phospho-Stat5, and cyclin D1, whereas combination treatment with 0.25 μM erlotinib and 3 μM γ-tocotrienol or 0.5 μM gefitinib and 3 μM γ-tocotrienol resulted in marked reduction in the levels of phospho-Stat3, phospho-Stat5, and cyclin D1 as compared to the vehicle-treated control group (Bachawal et al. 2010a). Furthermore, intracellular levels of phospho-Src, phospho-Stat1, Stat1, Stat3, and Stat5 were not found to differ greatly among any of the treatment groups (Bachawal et al. 2010a). These results demonstrated that combined low-dose treatment with γ-tocotrienol and erlotinib or gefitinib inhibited the activation of Stat3 and Stat5, but had no effect on the relative intracellular levels of phospho-Stat1 in +SA mammary tumor cells. Stat3 and Stat5 function to promote cell cycle progression and inhibit apoptosis, whereas Stat1 is considered to act as a tumor suppressor and its role in tumor progression and oncogenesis is not well understood and remains controversial (Bowman et al. 2000). Other studies show that Stats (particularly Stat3) can also be activated by Src, a non-receptor protein tyrosine kinase (Garcia et al. 2001; Olayioye et al. 1999). However, combined γ-tocotrienol and erlotinib or gefitinib treatment had no effect on the Src activation, suggesting that combined treatment–induced reductions in Stat3 phosphorylation are independent of Src activity (Bachawal et al. 2010a).
Treatment with 3 μM γ-tocotrienol, 0.25 μM erlotinib, or 0.5 μM gefitinib alone had no effect on the intracellular levels of phospho-PDK-1 and phospho-Akt, but combined treatment of 0.25 μM erlotinib with 3 μM γ-tocotrienol or 0.5 μM gefitinib with 3 μM γ-tocotrienol caused a large decrease in phospho-PDK-1 and phospho-Akt levels as compared to the vehicle-treated controls (Bachawal et al. 2010a). Total intracellular levels of PI3K p85 subunit, Akt, Pten, phospho-Pten, and PP2A catalytic subunit (active) showed no differences among any of the treatment groups. These observations suggest that combination treatment inhibits ErbB3 and ErbB4 receptor activity and subsequently blocks the recruitment of PI3K and further activation of Pdk-1 and Akt. It was also shown that combination treatment had no effect on the MAPK signaling in +SA mammary tumor cell. It is generally believed that EGF-dependent activation of MAPK signaling might primarily involve ErbB1 receptor activation, and previous studies have shown that γ-tocotrienol had no effect on ErbB1 receptor activation in this mammary tumor cell line (Sylvester and Shah 2005). In summary, the combination of γ-tocotrienol and erlotinib or gefitinib treatment inhibits the growth of mammary tumor cells specifically by suppressing the EGF-dependent Stat and Akt mitogenic signaling. These findings strongly suggest that tocotrienols used in combination with other agents that target ErbB receptors may enhance therapeutic responsiveness in breast cancer patients with aberrant EGF receptor signaling.
Conclusions
These studies demonstrate that combined treatment of γ-tocotrienol with statins or EGF receptor inhibitors results in a significantly enhanced and synergistic anticancer response against mammary tumor cells than that observed with high-dose monotherapy. Ideally, chemotherapy should destroy only cancer cells without harming normal cells. However, most traditional cancer chemotherapies are rather nonspecific in action and also damage normal cells, resulting in significant adverse and toxic side effects such as nausea, anemia, hair loss, fatigue, etc. Combination therapy provides an improved approach for limiting side effects and enhancing therapeutic outcome by utilizing drugs that target specific pathways that are vital to cancer cell growth and survival. Furthermore, cancer chemotherapy has been found to be more effective when drugs that work by different mechanisms are given in combination at lower optimal and synergistic doses. The synergistic effects of γ-tocotrienol and statins appear to result from the cooperative action of γ-tocotrienol to cause a down-regulation of HMGCoA reductase levels and statins to directly inhibit HMGCoA activity. This finding is of particular interest because this synergistic anticancer effect was observed using very low doses of each agent, suggesting that combined low-dose treatment of γ-tocotrienol with statins may have potential value in the treatment of breast cancer without causing myotoxicity that is associated with high-dose statin monotherapy. Similarly, combined treatment of γ-tocotrienol with receptor tyrosine kinase inhibitors synergistically inhibits the mammary tumor cell growth by inhibiting the activation of multiple EGF receptor family members and subsequently suppressing EGF-dependent Stat and Akt mitogenic signaling. These findings may also have significant clinical importance because tyrosine kinase inhibitors, such as erlotinib and gefitinib, target a single EGF receptor subtype, and these anticancer agents have shown disappointing results in the clinic when used as monotherapy. Again, experimental findings strongly suggest that tocotrienols used in combination with erlotinib or gefitinib target multiple EGF receptors and effectively block heterodimer cooperation and may have potential use to enhance therapeutic responsiveness in breast cancer patients with aberrant EGF receptor signaling.
References
American Cancer Society (2009) Cancer facts and figures. American Cancer Society Inc., Atlanta, GA
Arber N, Doki Y, Han EK, Sgambato A, Zhou P, Kim NH, Delohery T, Klein MG, Holt PR, Weinstein IB (1997) Antisense to cyclin D1 inhibits the growth and tumorigenicity of human colon cancer cells. Cancer Res 57:1569–1574
Arnold A, Papanikolaou A (2005) Cyclin D1 in breast cancer pathogenesis. J Clin Oncol 23:4215–4224
Bachawal SV, Wali VB, Sylvester PW (2010a) Combined gamma-tocotrienol and erlotinib/gefitinib treatment suppresses Stat and Akt signaling in murine mammary tumor cells. Anticancer Res 30:429–437
Bachawal SV, Wali VB, Sylvester PW (2010b) Enhanced antiproliferative and apoptotic response to combined treatment of gamma-tocotrienol with erlotinib or gefitinib in mammary tumor cells. BMC Cancer 10:84
Bartkova J, Lukas J, Muller H, Lutzhoft D, Strauss M, Bartek J (1994) Cyclin D1 protein expression and function in human breast cancer. Int J Cancer 57:353–361
Baserga R, Wiebel F (1969) The cell cycle of mammalian cells. Int Rev Exp Pathol 7:1–30
Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene 19:2474–2488
Britten CD (2004) Targeting ErbB receptor signaling: a pan-ErbB approach to cancer. Mol Cancer Ther 3:1335–1342
Brown MS, Faust JR, Goldstein JL, Kaneko I, Endo A (1978) Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J Biol Chem 253:1121–1128
Caldon CE, Daly RJ, Sutherland RL, Musgrove EA (2006) Cell cycle control in breast cancer cells. J Cell Biochem 97:261–274
Campbell MJ, Esserman LJ, Zhou Y, Shoemaker M, Lobo M, Borman E, Baehner F, Kumar AS, Adduci K, Marx C, Petricoin EF, Liotta LA, Winters M, Benz S, Benz CC (2006) Breast cancer growth prevention by statins. Cancer Res 66:8707–8714
Carraway KL 3rd, Cantley LC (1994) A neu acquaintance for erbB3 and erbB4: a role for receptor heterodimerization in growth signaling. Cell 78:5–8
Chan KK, Oza AM, Siu LL (2003) The statins as anticancer agents. Clin Cancer Res 9:10–19
Coleman ML, Marshall CJ, Olson MF (2004) RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat Rev Mol Cell Biol 5:355–366
Cottrell RC (1991) Introduction: nutritional aspects of palm oil. Am J Clin Nutr 53:989S–1009S
Downward J (1998) Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol 10:262–267
Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, Cox CE, Falcone R, Fairclough R, Parsons S, Laudano A, Gazit A, Levitzki A, Kraker A, Jove R (2001) Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 20:2499–2513
Gerson RJ, MacDonald JS, Alberts AW, Kornbrust DJ, Majka JA, Stubbs RJ, Bokelman DL (1989) Animal safety and toxicology of simvastatin and related hydroxy-methylglutaryl-coenzyme A reductase inhibitors. Am J Med 87:28S–38S
Goh SH, Hew NF, Norhanom AW, Yadav M (1994) Inhibition of tumour promotion by various palm-oil tocotrienols. Int J Cancer 57:529–531
Goldstein JL, Brown MS (1990) Regulation of the mevalonate pathway. Nature 343:425–430
Goldstein JL, DeBose-Boyd RA, Brown MS (2006) Protein sensors for membrane sterols. Cell 124:35–46
Graaf MR, Richel DJ, van Noorden CJ, Guchelaar HJ (2004) Effects of statins and farnesyltransferase inhibitors on the development and progression of cancer. Cancer Treat Rev 30:609–641
Grillo M, Bott MJ, Khandke N, McGinnis JP, Miranda M, Meyyappan M, Rosfjord EC, Rabindran SK (2006) Validation of cyclin D1/CDK4 as an anticancer drug target in MCF-7 breast cancer cells: effect of regulated overexpression of cyclin D1 and siRNA-mediated inhibition of endogenous cyclin D1 and CDK4 expression. Breast Cancer Res Treat 95:185–194
Hall M, Peters G (1996) Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res 68:67–108
Hermes M, Schormann W, Brulport M, Uhlemann K, Lupatsch F, Horn LC, Schumann A, Allgaier C, Weishaupt M, Engeland K, Muller GA, Mossner J, Bauer A, Schiffer IB, Gebhard S, Schmidt M, Lausch E, Prawitt D, Wilhelm C, Hengstler JG (2008) Trastuzumab therapy vs tetracycline controlled ERBB2 downregulation: influence on tumour development in an ERBB2-dependent mouse tumour model. Br J Cancer 98:1525–1532
Karunagaran D, Tzahar E, Beerli RR, Chen X, Graus-Porta D, Ratzkin BJ, Seger R, Hynes NE, Yarden Y (1996) ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: implications for breast cancer. EMBO J 15:254–264
Knowlden JM, Gee JM, Seery LT, Farrow L, Gullick WJ, Ellis IO, Blamey RW, Robertson JF, Nicholson RI (1998) c-erbB3 and c-erbB4 expression is a feature of the endocrine responsive phenotype in clinical breast cancer. Oncogene 17:1949–1957
Kornbrust DJ, MacDonald JS, Peter CP, Duchai DM, Stubbs RJ, Germershausen JI, Alberts AW (1989) Toxicity of the HMG-coenzyme A reductase inhibitor, lovastatin, to rabbits. J Pharmacol Exp Ther 248:498–505
Liang J, Slingerland JM (2003) Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2:339–345
Ling YH, Lin R, Perez-Soler R (2008) Erlotinib induces mitochondrial-mediated apoptosis in human H3255 non-small-cell lung cancer cells with epidermal growth factor receptorL858R mutation through mitochondrial oxidative phosphorylation-dependent activation of BAX and BAK. Mol Pharmacol 74:793–806
Maltese WA, Defendini R, Green RA, Sheridan KM, Donley DK (1985) Suppression of murine neuroblastoma growth in vivo by mevinolin, a competitive inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Clin Invest 76:1748–1754
Mantha AJ, Hanson JE, Goss G, Lagarde AE, Lorimer IA, Dimitroulakos J (2005) Targeting the mevalonate pathway inhibits the function of the epidermal growth factor receptor. Clin Cancer Res 11:2398–2407
Marshall E (1993) Epidemiology. Search for a killer: focus shifts from fat to hormones [news] [see comments]. Science 259:618–621
Massague J (2004) G1 cell-cycle control and cancer. Nature 432:298–306
Massague J, Pandiella A (1993) Membrane-anchored growth factors. Annu Rev Biochem 62:515–541
Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY (1994) D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol 14:2066–2076
McIntyre BS, Briski KP, Gapor A, Sylvester PW (2000a) Antiproliferative and apoptotic effects of tocopherols and tocotrienols on preneoplastic and neoplastic mouse mammary epithelial cells. Proc Soc Exp Biol Med 224:292–301
McIntyre BS, Briski KP, Tirmenstein MA, Fariss MW, Gapor A, Sylvester PW (2000b) Antiproliferative and apoptotic effects of tocopherols and tocotrienols on normal mouse mammary epithelial cells. Lipids 35:171–180
Medina D (1974) Mammary tumorigenesis in chemical carcinogen-treated mice. II. Dependence on hormone stimulation for tumorigenesis. J Natl Cancer Inst 53:223–226
Mo H, Elson CE (2004) Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp Biol Med (Maywood) 229:567–585
Nagasawa H, Yanai R, Taniguchi H (1976) Importance of mammary gland DNA synthesis on carcinogen-induced mammary tumorigenesis in rats. Cancer Res 36:2223–2226
Nakanishi M, Goldstein JL, Brown MS (1988) Multivalent control of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J Biol Chem 263:8929–8937
Nakayama M, Ju HR, Sugano M, Hirose N, Ueki T, Doi F, Eynard AR (1993) Effect of dietary fat and cholesterol on dimethylbenz[a]-anthracene-induced mammary tumorigenesis in Sprague-Dawley rats. Anticancer Res 13:691–698
Nandi S, Guzman RC, Yang J (1995) Hormones and mammary carcinogenesis in mice, rats, and humans: a unifying hypothesis. Proc Natl Acad Sci USA 92:3650–3657
Norbury C, Nurse P (1992) Animal cell cycles and their control. Annu Rev Biochem 61:441–470
Normanno N, Campiglio M, De LA, Somenzi G, Maiello M, Ciardiello F, Gianni L, Salomon DS, Menard S (2002) Cooperative inhibitory effect of ZD1839 (Iressa) in combination with trastuzumab (Herceptin) on human breast cancer cell growth. Ann Oncol 13:65–72
Normanno N, Bianco C, De Luca A, Maiello MR, Salomon DS (2003) Target-based agents against ErbB receptors and their ligands: a novel approach to cancer treatment. Endocr Relat Cancer 10:1–21
Okubo S, Kurebayashi J, Otsuki T, Yamamoto Y, Tanaka K, Sonoo H (2004) Additive antitumour effect of the epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (Iressa, ZD1839) and the antioestrogen fulvestrant (Faslodex, ICI 182, 780) in breast cancer cells. Br J Cancer 90:236–244
Olayioye MA, Beuvink I, Horsch K, Daly JM, Hynes NE (1999) ErbB receptor-induced activation of stat transcription factors is mediated by Src tyrosine kinases. J Biol Chem 274:17209–17218
Olayioye MA, Neve RM, Lane HA, Hynes NE (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J 19:3159–3167
Parker RA, Pearce BC, Clark RW, Gordon DA, Wright JJ (1993) Tocotrienols regulate cholesterol production in mammalian cells by post-transcriptional suppression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem 268:11230–11238
Rogers AE, Lee SY (1986) Chemically-induced mammary gland tumors in rats: modulation by dietary fat. Prog Clin Biol Res 222:255–282
Russo IH, Russo J (1978) Developmental stage of the rat mammary gland as determinant of its susceptibility to 7, 12-dimethylbenz[a]anthracene. J Natl Cancer Inst 61:1439–1449
Samant GV, Sylvester PW (2006) gamma-Tocotrienol inhibits ErbB3-dependent PI3K/Akt mitogenic signalling in neoplastic mammary epithelial cells. Cell Prolif 39:563–574
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225
Sears RC, Nevins JR (2002) Signaling networks that link cell proliferation and cell fate. J Biol Chem 277:11617–11620
Seeger H, Wallwiener D, Mueck AO (2003) Statins can inhibit proliferation of human breast cancer cells in vitro. Exp Clin Endocrinol Diabetes 111:47–48
Shah SJ, Sylvester PW (2005) Gamma-tocotrienol inhibits neoplastic mammary epithelial cell proliferation by decreasing Akt and nuclear factor kappaB activity. Exp Biol Med 230:235–241
Shapiro GI (2006) Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 24:1770–1783
Sherr CJ (1993) Mammalian G1 cyclins. Cell 73:1059–1065
Shibata MA, Kavanaugh C, Shibata E, Abe H, Nguyen P, Otsuki Y, Trepel JB, Green JE (2003) Comparative effects of lovastatin on mammary and prostate oncogenesis in transgenic mouse models. Carcinogenesis 24:453–459
Shibata MA, Ito Y, Morimoto J, Otsuki Y (2004) Lovastatin inhibits tumor growth and lung metastasis in mouse mammary carcinoma model: a p53-independent mitochondrial-mediated apoptotic mechanism. Carcinogenesis 25:1887–1898
Shirode AB, Sylvester PW (2010) Synergistic anticancer effects of combined gamma-tocotrienol and celecoxib treatment are associated with suppression in Akt and NFkappaB signaling. Biomed Pharmacother 64:327–332
Sundram K, Khor HT, Ong AS (1990) Effect of dietary palm oil and its fractions on rat plasma and high density lipoprotein lipids. Lipids 25:187–193
Sylvester PW, Shah SJ (2005) Mechanisms mediating the antiproliferative and apoptotic effects of vitamin E in mammary cancer cells. Front Biosci 10:699–709
Sylvester PW, Theriault A (2003) Role of tocotrienols in the prevention of cardiovascular disease and breast cancer. Curr Top Nutraceutical Res 1:121–136
Sylvester PW, Aylsworth CF, Van Vugt DA, Meites J (1983) Effects of alterations in early hormonal environment on development and hormone dependency of carcinogen-induced mammary tumors in rats. Cancer Res 43:5342–5346
Sylvester PW, Ip C, Ip MM (1986a) Effects of high dietary fat on the growth and development of ovarian-independent carcinogen-induced mammary tumors in rats. Cancer Res 46:763–769
Sylvester PW, Russell M, Ip MM, Ip C (1986b) Comparative effects of different animal and vegetable fats fed before and during carcinogen administration on mammary tumorigenesis, sexual maturation, and endocrine function in rats. Cancer Res 46:757–762
Thibault A, Samid D, Tompkins AC, Figg WD, Cooper MR, Hohl RJ, Trepel J, Liang B, Patronas N, Venzon DJ, Reed E, Myers CE (1996) Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res 2:483–491
Toker A (2000) Protein kinases as mediators of phosphoinositide 3-kinase signaling. Mol Pharmacol 57:652–658
Torii S, Yamamoto T, Tsuchiya Y, Nishida E (2006) ERK MAP kinase in G cell cycle progression and cancer. Cancer Sci 97:697–702
Tran H, Brunet A, Griffith EC, Greenberg ME (2003) The many forks in FOXO’s road. Sci STKE 2003:RE5
Tzahar E, Pinkas-Kramarski R, Moyer JD, Klapper LN, Alroy I, Levkowitz G, Shelly M, Henis S, Eisenstein M, Ratzkin BJ, Sela M, Andrews GC, Yarden Y (1997) Bivalence of EGF-like ligands drives the ErbB signaling network. EMBO J 16:4938–4950
Vogel VG (1993) Breast cancer risk: weaving facts into fabric. Breast Cancer Res Treat 28:91–96
Wali VB, Sylvester PW (2007) Synergistic antiproliferative effects of gamma-tocotrienol and statin treatment on mammary tumor cells. Lipids 42:1113–1123
Wali VB, Bachawal SV, Sylvester PW (2009a) Suppression in mevalonate synthesis mediates antitumor effects of combined statin and gamma-tocotrienol treatment. Lipids 44:925–934
Wali VB, Bachawal SV, Sylvester PW (2009b) Combined treatment of gamma-tocotrienol with statins induce mammary tumor cell cycle arrest in G1. Exp Biol Med (Maywood) 234:639–650
Yarden Y (2001) Biology of HER2 and its importance in breast cancer. Oncology 61(Suppl 2):1–13
Acknowledgments
This work was performed in the College of Pharmacy at the University of Louisiana at Monroe, Monroe, LA and supported in part by grants from the National Institutes of Health (Grant CA 86833) and First Tech International Ltd. The authors would also like to thank the Malaysian Palm Oil Board (MPOB) and Carotech Bhd for their support in generously supplying γ-tocotrienol for use in these studies.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is part of a Special Issue entitled “Tocotrienols in health and disease” (Guest editors: Bharat B. Aggarwal and Kalaniti Nesaretnam).
Rights and permissions
About this article
Cite this article
Sylvester, P.W. Synergistic anticancer effects of combined γ-tocotrienol with statin or receptor tyrosine kinase inhibitor treatment. Genes Nutr 7, 63–74 (2012). https://doi.org/10.1007/s12263-011-0225-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12263-011-0225-y