
Abstract
Alzheimer's disease (AD) involves a complex pathological cascade thought to be initially triggered by the accumulation of β-amyloid (Aβ) peptide aggregates or aberrant amyloid precursor protein (APP) processing. Much is known of the factors initiating the disease process decades prior to the onset of cognitive deficits, but an unclear understanding of events immediately preceding and precipitating cognitive decline is a major factor limiting the rapid development of adequate prevention and treatment strategies. Multiple pathways are known to contribute to cognitive deficits by disruption of neuronal signal transduction pathways involved in memory. These pathways are altered by aberrant signaling, inflammation, oxidative damage, tau pathology, neuron loss, and synapse loss. We need to develop stage-specific interventions that not only block causal events in pathogenesis (aberrant tau phosphorylation, Aβ production and accumulation, and oxidative damage), but also address damage from these pathways that will not be reversed by targeting prodromal pathways. This approach would not only focus on blocking early events in pathogenesis, but also adequately correct for loss of synapses, substrates for neuroprotective pathways (e.g., docosahexaenoic acid), defects in energy metabolism, and adverse consequences of inappropriate compensatory responses (aberrant sprouting). Monotherapy targeting early single steps in this complicated cascade may explain disappointments in trials with agents inhibiting production, clearance, or aggregation of the initiating Aβ peptide or its aggregates. Both plaque and tangle pathogenesis have already reached AD levels in the more vulnerable brain regions during the “prodromal” period prior to conversion to “mild cognitive impairment (MCI).” Furthermore, many of the pathological events are no longer proceeding in series, but are going on in parallel. By the MCI stage, we stand a greater chance of success by considering pleiotropic drugs or cocktails that can independently limit the parallel steps of the AD cascade at all stages, but that do not completely inhibit the constitutive normal functions of these pathways. Based on this hypothesis, efforts in our laboratories have focused on the pleiotropic activities of omega-3 fatty acids and the anti-inflammatory, antioxidant, and anti-amyloid activity of curcumin in multiple models that cover many steps of the AD pathogenic cascade (Cole and Frautschy, Alzheimers Dement 2:284–286, 2006).
Similar content being viewed by others


Background
Alzheimer Pathogenesis
Genetic data from Down's syndrome and early-onset Alzheimer's disease (AD) families argue that increased production of a 42-amino-acid β-amyloid peptide (Aβ42) from birth is sufficient to cause clinical onset of AD four to five decades later. Cross-sectional investigations of pathology and more recent biomarker and imaging studies show that AD pathogenesis typically begins with nascent Aβ42 aggregates and deposits and related tau accumulation in vulnerable brain regions, for example, medial temporal regions including entorhinal cortex and hippocampus. The early region-specific accumulation of diffuse Aβ and tau eventually matures to plaques and tangles. These lead to inflammatory glial cells that emerge in many vulnerable brain regions with distinct differences in the activation pattern for tangles and plaques. Ultimately, after decades of pathology buildup in a prolonged “prodromal” phase, synaptic deficits and neuron loss appear to play a major role in driving cognitive decline. Supporting this, cross-sectional autopsy reports have identified individuals who are seemingly cognitively normal but with high levels of plaques [2] and or tangle pathology, consistent with the notion that much of the pathology occurs preceding rather than in parallel with cognitive decline [3, 4]. During the clinically relevant cognitive decline stages, deficits are associated with continued spreading of tangles, synaptic impairments, and neuron loss. One sensitive biomarker, uses the amyloid binding of radiolabelled Pittsburgh compound-B (PiB) for positron emission tomography (PET) neuroimaging has proven to be fairly sensitive to detect amyloid or its correlates, apparently at pre-mild cognitive impairment (MCI) stages. Alteration of some CSF biomarkers is fairly predictive of decline, particularly an increase in tau to Aβ42 ratios. MRI hippocampal volumetrics have been effective in tracking regional defects in neurodegenerative tracts and fluorodeoxyglucose (FDG) PET in detecting region-dependent hypometabolism in glucose metabolism. These changes support a model where amyloid accumulation occurs in the prodromal period preceding clinical onset, followed by a progression of tau pathology and neurodegeneration closely related to cognitive decline [5]. This conclusion is supported by earlier cross-sectional studies showing a close correlation between cognitive decline and synapse loss but poorer correlations with tau or Aβ pathology [6].
Need for Refinement of the Alzheimer's Cascade Hypothesis
Amyloid plaques have been hypothesized to play a major role in pathogenesis since their description by Alois Alzheimer even before the Aβ peptide was sequenced in 1984 by George Glenner. The amyloid cascade hypothesis evolved primarily from the genetic data on early-onset AD mutations that increase Aβ42 production leading to its aggregation combined with evidence that Aβ42 aggregates can initiate a cascade of pathology found in AD [7]. Supporting the Aβ42 status as a cause or initiator of AD, Aβ was shown to accumulate very early in the disease process and reached AD levels while patients are still cognitively intact [8]. Because there is compelling evidence that mutations that cause elevated Aβ42 production and accumulation are sufficient to cause AD, it was inferred that effective targeting Aβ42 early enough should prevent the disease. The amyloid cascade hypothesis, officially defined by John Hardy in 1992 [9], was challenged by Robert D. Terry and colleagues Robert Katzman and E. Masliah [6] who noted that cognitive loss correlated well with synapse loss, but not so well with tangles and poorly with Aβ deposited as plaques. They also pointed out many cases of “high plaque” cognitively normal individuals, arguing that Aβ42 accumulation was not sufficient to cause AD. Since some of the initial clinical trials directed at Aβ peptide have not met expectations for robust treatment effects, the causal role for Aβ continues to be challenged. Strong opponents of the hypothesis now include Mark Smith and George Perry, who have criticized the field as being too “amyloidocentric” [10, 11], emphasized oxidative damage and cited data reporting amyloid precursor protein (APP) [12] or Aβ enhancing synaptic plasticity [13]. While we and others agree that the impact of Aβ aggregates on memory in AD patients is not direct since the prodromal period of Aβ aggregate accumulation is decades long, we argue that the evidence that Aβ precipitates the disease process remains compelling because the implicated pathways in the amyloid cascade hypothesis, such as oxidative damage, are not necessarily reversible by late intervention [14]. A recent review of the hypothesis states that Aβ “causality has been neither proved nor disproven” [15]. Further refinement of an Alzheimer's cascade, amyloid or otherwise, may improve trial outcomes by timing interventions to incorporate what we know about stages, lagging effects, and the reversibility of different pathways [5].
For example, we now know from trial data that antagonizing amyloid or its oligomers well after their accumulation with vaccine is not very unlikely to reverse the clinical symptoms of disease. Agents like R-fluribiprofen (Tarenflurbil or Flurizan, Myriad Pharmaceuticals) that lower Aβ production [16] have shown promise in phase II [17] trials but very clearly failed in phase III [18] trials. Several other anti-Aβ agents have also failed in trials and, while none of these have proven that they lowered amyloid in vivo, the active vaccine and passive immunization trials have shown that they do, using autopsy and PiB, but whatever the clinical benefit achieved, the patients clearly remained demented. While not by any means a disproof of the amyloid cascade hypothesis, these results argue that anti-Aβ therapies are not likely to be sufficient for the treatment of established AD after Aβ aggregates have initiated the cascade. There are many analogous situations. For example, cancer researchers know very well that genetic defects or carcinogens induce mutations in specific oncogenes and tumor suppressor genes to cause different cancers, but they find little or no utility in treating established tumors with anti-carcinogens. In fact, many cancer chemoprevention efforts with anti-carcinogens have failed despite strong evidence for efficacy in animal models where most interventions were able to act at the initial mutation stages of carcinogenesis rather than the subsequent tumor promotion phases. Obviously, this says nothing about the validity of the well-established theory that mutagens cause cancer.
The majority of preclinical research targeting Aβ accumulation has been implemented using animal models lacking the neurodegenerative phase, and therefore, the tested interventions act prior to or during the amyloid accumulation phase and not during the missing major neurodegenerative phase. Models with mutant APP should be viewed as most useful for the initial amyloid accumulation segment of prodromal AD and thus most relevant for predicting primary prevention trial results, but not for selecting agents for clinical trials with established AD. The interventions directed against Aβ as an initiating factor are all predicted to work much better when and if they are used for prevention. However, at preclinical stages, therapies targeting only Aβ have little or no opportunity to show clinically relevant therapeutic benefits to offset their risks of side effects. One corollary is that the best anti-Aβ interventions should have very strong safety records, which raises the bar for new drugs for prevention. This reasoning has been a major factor that has pushed our group to focus primarily on anti-amyloid interventions with a long history of use and favorable known safety profiles including ibuprofen, curcumin, and the omega-3 fatty acid, docosahexaenoic acid (DHA).
A second conclusion from this line of reasoning is that interventions using models driven by Aβ that do not progress to extensive tau pathology and neurodegeneration are unlikely to predict success in trials where Aβ, tau pathology, and major neuron loss are already present, e.g., in MCI. The rodent models of Aβ pathology also fail to develop the same level of inflammatory response [19]. In some models, cognitive deficits emerge prior to plaque deposition when Aβ first forms oligomers; these oligomers are sufficient to cause cognitive deficits in the absence of tau/tangle pathology or chronic inflammation [20]. Aβ oligomers cause defects in long-term potentiation (LTP) linked to memory deficits [21]. However, an early increase in cognitive deficits with rising Aβ aggregates is not the situation with humans where plaques and Aβ oligomers are formed many years and in fact decades prior to identifiable and clinically relevant impairment. Furthermore, Aβ is not the only factor capable of causing cognitive decline. Inflammation and elevated inflammatory cytokines can be sufficient to cause defects in LTP [22] and cognition [23].
Similarly, cognitive deficits associated with tau pathology and neurodegeneration in humans with certain forms of frontal temporal dementia, in animal models lacking Aβ accumulation [24, 25], and even in animals lacking tangles but with hyperphosphorylated tau suggest that tau defects in the absence of Aβ aggregates are sufficient to cause cognitive deficits [26]. Clearly, treatments that fail to control inflammation or tau-mediated deficits are incomplete. Tau pathology, neuron loss, and dementia persist after late removal of Aβ. Therefore, with either MCI or AD patients, one should never have expected the same level of clinical benefits from targeting Aβ as seen in animal models with only limited neuroinflammation and tau pathology and early intervention. The efficacy of reducing one or more pools of accumulated or nascent Aβ by any of the many anti-Aβ therapies can only be answered by ongoing clinical trials. However, the initial results with anti-Aβ vaccine strongly suggest only a partial efficacy with persistent tau and vascular pathology and relatively unabated further cognitive decline [27, 28].
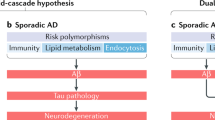
In agreement with a recent commentary, it seems clear that targeting amyloid may work at some very early stage of AD, but is probably not sufficient to halt the pathological cascade at later stages [29]. Because the entire cascade proceeds at different rates in different brain regions, each pathological stage may be occurring in at least one brain region at the same time when interventions begin beyond the earliest “Braak stages” of pathogenesis [30]. This means that, while some clinical benefits are likely to be obtained at all stages, a growing recognition provides a strong rationale for a pleiotropic approach targeting the cascade at multiple levels combining therapy for Aβ, inflammation, oxidative damage, tau pathology, and neuroprotection. Originally conceptual and theoretical, the “amyloid cascade hypothesis” has now taken many forms as new details emerge. Some of the controversies could be resolved and tested if the cascade hypothesis was refined to deal with specific stages. We argue that rather than one model to recapitulate the complexity of AD, we need different animal models to appropriately dissect out different segments of the cascade to develop better stage-dependent therapies (Figs. 1 and 2).

Stage-dependent pathogenesis: This figure illustrates the complexity of different pathogenic events (inflammation, synaptic loss, oxidative damage in addition to imaging, and CSF biomarkers) at different stages, showing that there is extensive tau and Aβ pathology prior to symptoms. Identifying plasma or CSF biomarkers specific for targeted pathway (inflammation, neurodegeneration) is important for the rapid development of treatments and early interventions

Hypothetical Alzheimer cascade: Argument for pleiotropic targeting. This schematic diagram illustrates a refinement of the amyloid cascade, suggesting that more than one pathogenic pathway contributes to memory loss and demonstrating how anti-Aβ or other monotherapies would not succeed on their own. Examples of such pathogenic pathways that need to be targeted are oxidative damage (which depletes membrane n-3 fatty acids like DHA), aberrant inflammation, and pTau accumulation. In agreement with the “baptists,” Aβ accumulation is a triggering event. But in agreement with the “taoists,” both tau aggregates and increased pTau are likely to be major contributors to memory deficits (even though tangle pathology is not specific to AD). Nevertheless, reducing tau aggregation is unlikely to be sufficient since other events including impaired energy metabolism and aberrant are contributing to memory deficits. Compounds like curcumin or DHA or both are likely to be effective in targeting multiple steps
Mechanisms
Aβ and Tau
Aβ aggregates, notably Aβ oligomers, can induce inflammation, oxidative damage, and multiple signal transduction events including activation of tau kinases [31]. The role of tau pathology is progressive and profound with roles for both gain of toxic functions from tau aggregates [32] as well as a loss of normal tau function [33, 34]. Strategies for dealing with tau pathology include tau aggregation inhibitors [35], drugs that compensate for loss of tau function by stabilizing microtubules, albeit with some toxicity issues [36], and inhibitors of tau kinases such as glycogen synthase kinase-3β (GSK3β) [37]. Unlike other tau kinases, GSK3β has a relatively high constitutive activity that plays an important normal function in long-term depression and cognition. GSK3β's activity is not clearly elevated in AD brain even though it is one mediator of acute Aβ toxicity and this questions the potential of GSK3β as an optimal therapeutic target [38]. As with Aβ, the most appealing strategy is to target the abnormal tau aggregates, but this alone may not be sufficient if loss of tau function is caused by hyperphosphorylation which is central to many deficits [34]. Again, a pleiotropic treatment that mitigates both tau hyperphosphorylation and the loss of tau function and also toxic tau aggregates is more likely to succeed on the multiple fronts we are fighting.
Oxidative Damage
There is abundant evidence for oxidative damage to proteins, lipids, and DNA in the brains of AD patients [39–42]. There may be some causal role related to amyloid because amyloid-induced neurotoxicity can be reduced with vitamin E and other antioxidants [43, 44]. Vitamin E can reduce amyloid accumulation at early but not late stages in APP transgenic mice [45]. Similarly, in another APP transgenic model, the antioxidant R-lipoic acid effectively reduced oxidative damage, but did not attenuate Aβ pathogenesis or correct cognitive deficits [46]. Thus, even though oxidative stress can increase Aβ production via effects on BACE1, inhibiting oxidation (and presumably Aβ production), targeting oxidative damage alone does not adequately prevent Aβ accumulation and does not appear sufficient to protect cognition. That said, one small AD trial with α-lipoate in combination with DHA was reported effective in preventing cognitive decline, but not sufficiently powered to be compelling [47]. Oxidative stress from aging, Aβ, or metals may also be a cause of kinase activation related to tau hyperphosphorylation and cell cycle reactivation as implicated in neurodegeneration and AD [48]. Some of the oxidative damage may be linked to inflammation and eicosanoid production [49]. Epidemiological studies have also shown some support for reduced risk with higher antioxidant intake, including vitamin E [50, 51]. However, vitamin E (α-tocopherol) has had limited efficacy with established AD [52] or MCI [53]. Unfortunately, these large trials did not monitor biomarkers to prove that the treatments effectively reduced oxidative damage. In particular, one outcome shows that excessive α-tocopherol causes depletion of endogenous γ-tocopherol and has limited antioxidant effects on nitrate radicals [54]. Ongoing analysis of a multi-site clinical trial with an antioxidant cocktail will better address this issue. However, based on current evidence, we conclude that monotherapy to reduce oxidative damage may be partially neuroprotective, but since it does not reduce inflammation or other pathogenic cascades, it is probably not sufficient to reduce Aβ accumulation or to slow progression in late stages.
Aberrant Inflammation Impedes Clearance and Exacerbates Pathogenesis
There is strong evidence of neuroinflammatory changes in AD patients, and these changes are independent of tau and Aβ pathogenesis and can cause cognitive, neurodegenerative, or oxidative damage [55, 56]. One of the most well-characterized neuroinflammatory responses in AD has been increases in interleukin-1. For example, IL-1β can exacerbate Aβ pathogenesis via increasing β-secretase (BACE) [57–60]. Even 14 days overexpression of IL-1β in the adult mouse can impair acquisition and retention of spatial learning [61]. IL-1β appears to mediate impairment of memory consolidation after infection [62] and can also exacerbate tau pathogenesis, probably via its impact on tau kinases [63]. In fact, Alzheimer's patients have microglial dystrophy that is associated with tau pathology, and this change is not observed in AD animal models. This is consistent for a role of microglia and inflammatory cytokines in tau pathogenesis.
Some inflammatory responses that contribute to pathogenesis may have opposing (beneficial) roles in reducing the Aβ burden in some paradigms. One example is C1q, a complement protein, which can stimulate phagocytosis and clearance of amyloid [64]. Consistent with this idea, inhibiting C3 activation by expressing soluble complement receptor-related protein Y (sCrry) was shown to exacerbate pathogenesis in the AD model [65]. Similarly, IL-1β can reduce plaque pathogenesis in some paradigms [66], but ultimately the responses are associated with the disease process, suggesting that there is dysfunction signaling in these pathways. Thus, increasing complement activation and IL-1β would likely be poor therapeutic targets with net adverse effects in AD, not just because it would not restore the signaling defects, but because of the adverse effects on complement activation cannot be modeled well in animals.
Another important factor is that aberrant inflammatory responses observed in AD are much more robust and extensive [67] than can be observed in APP transgenic animal models where inflammation is blunted and focal [68, 69]. Thus, caution must be made in direct translation of proinflammatory therapeutics (vaccines) using animal models, which likely underestimate the potential for adverse inflammatory effects in AD patients who have much more robust complement activation. Passive or active Aβ vaccines, which can clear amyloid from the brain in animals and humans, appear to be most beneficial for cognition in animal models if interventions are pre- or early pathological stage [70]. Late intervention vaccine treatment actually failed to improve cognition in animal models [71] and the vaccines in human trials have so far failed to yield major improvements in cognition. Late intervention in another animal model, aging canines, did not yield major improvements in executive function [71], suggesting that active vaccination may need to start before or very early in the development of clinically significant AD-related pathology. The role of neuroinflammation in AD is complex, justifying the title of a recent review “Neuroinflammation in Alzheimer's disease and mild cognitive impairment: a field in its infancy” [72]. How to resolve aberrant inflammation contributing to pathogenesis without disrupting beneficial inflammation is a key problem in developing therapeutics. Another key problem is the failure of many translational animal model studies to focus on late preclinical intervention (post-pathology) studies, which (contrary to early preclinical intervention studies) have generally not predicted efficacy in clinical trials. The obvious reason why many animal model studies employing early intervention (pre-pathology) will end in clinical trial failures is that the models are only relevant to interventions in very early stages (age 40s, 50s, or 60s). There are also other overlooked differences between animal models and AD. Unlike the animal models, all components of the complement cascade required for complement activation and perforation of membranes are upregulated in AD [72, 73]. In fact, despite the beneficial effects of C1q in Aβ clearance in mouse models, aggregated Aβ can directly activate the complement cascade [74]. Since factors associated with complement activation (C1q) can also play a role in clearance, the precise molecular target to reduce complement-related neuron damage (that will not impede clearance) needs to be identified.
Another downside of chronic overexpression of IL-1 is that it interferes with the normal regulation of the hypothalamic–pituitary–adrenal (HPA) axis, resulting in inappropriate elevation of glucocorticoids [75]. This is supported by the observation that many Alzheimer's patients exhibit hypercortisolism and impaired regulation of the HPA axis that cannot be solely explained by damage in the hippocampus [55]. While slightly elevated glucocorticoids can have beneficial effects, such as in caloric restriction, abnormally high glucocorticoids that occur with chronic stress and in AD may contribute to energy deficits [76] and insulin resistance [77], a risk factor for AD [78].
Interestingly, inhibiting brain inflammation with ibuprofen fails to correct downstream oxidative damage [79]. This could either indicate that oxidative damage caused by chronic inflammation is irreversible or that factors in addition to inflammation also contribute to oxidative damage. This is another rationale for a pleiotropic drug or a cocktail that controls, at least, both inflammation and oxidative damage. One common misperception is that inhibiting inflammation with non-steroidal anti-inflammatory drugs (NSAIDs) may impede phagocytic clearance, leading to increased accumulation of Aβ, but in fact inhibiting inflammation actually tends to reduce Aβ burden. Thus, it is feasible that vaccines and NSAIDs could even work in synergism, a yin yang approach. Since more than one factor can cause inflammation (oxidative damage, Aβ, or even tauopathy-induced neurodegeneration) and all are in play prior to the earliest onset of MCI, antagonizing one of the above alone is unlikely to reverse the adverse effect of inflammation. Finally, despite compensatory responses present in both AD models and in AD, there is an intermediate state of aberrant inflammation or partial microglial activation that contributes to both impaired phagocytic clearance of Aβ and activation of pathogenic cascades that likely contribute to the progression of disease.
Are NSAIDs Pleiotropic and Stage-Specific in AD Pathogenesis?
Chronic NSAID consumption has been consistently associated with reduced risk for AD, suggesting that conventional NSAIDs that target the cyclooxygenase (COX) enzymes for AD interventions [80]. The most recent epidemiology continues to support risk reduction for conversion to MCI from chronic naproxen or ibuprofen usage for at least 2 years duration [81]. However, the results of treatment trials to date with many NSAIDs, including COX-2 inhibitor and naproxen, suggest little efficacy with shorter-term treatment (<2 years) or even aggravation of symptoms with established AD [82]. Furthermore, with initial results from a prevention trial (Alzheimer's Disease Anti-inflammatory Prevention Trial [ADAPT]) that was halted over safety concerns, neither naproxen nor COX-2 inhibitor was protective after 24 months [83]. Intriguingly, after halting of the trial but continuing with follow-up, it was discovered that some patients entering the trial had cognitive deficits and should have been excluded, and when these patients were excluded, and patients followed up (after being off of naproxen for 1.5 years), there was a long-term protective benefit in the naproxen group with an apparently significant reduction in conversion to AD as well as 40% reduction of CSF tau/Aβ42 in non-demented naproxen users, a biomarker predicting decline [84]. Together with NSAID epidemiology data showing risk reduction with early (pre-symptomatic) intake with naproxen and ibuprofen [81, 85], the new ADAPT trial results support the potential of NSAID prevention but raise caution for aggravation of symptoms if intervention is in an early post-symptomatic stage. We and others have found that chronic ibuprofen treatment reduces amyloid accumulation [59, 86, 87] and Aβ-induced tau [88], which is probably the simplest logical mechanism explaining the impact on CSF tau and Aβ42 in the ADAPT trial. Because the trial was halted over safety concerns, possible disease-modifying prevention also strongly encourages the search for alternative NSAIDs with greater safety and equal or greater efficacy. Furthermore, if NSAIDs prove to be effective in AD, it may be only effective in ApoE4 (e.g., a pharmacogenomic efficacy) as one small trial found that a subsample of genotyped patients, ApoE epsilon4 carriers treated with ibuprofen (n = 27), were the only group without significant cognitive decline, while in non-ApoE4 carriers, ibuprofen had no impact [89]. Similar genotype effects have been reported in epidemiological studies.
Traditional NSAIDs typically inhibit both COX-1 and COX-2, enzymes that metabolize arachidonic acid (AA) after it is freed from membrane phospholipids by phospholipases. For example, while COX-2 is inducible and elevated in neurons early in AD, COX-1 is constitutive but most likely the better target for reactive microglia and inflammation [90]. Beyond the role of NSAIDs in reducing COX metabolites of AA, there is now evidence that AA and its non-COX metabolites are also directly relevant to AD. A recent lipidomic study showed that cytosolic phospholipase A2 (cPLA2; GIVA PLA2), a phospholipase that releases AA from phospholipids, is activated in AD and AD model mice, resulting in elevations of all of AA's different COX and lipoxygenase (LOX) metabolites. Knockout of cPLA2 protected from cognitive deficits in the AD transgenic mice [91].
Another novel mechanism of Aβ is its ability to activate NADPH oxidase and induce the production of reactive oxygen species (ROS) in astrocytes and neurons. In turn, ROS, depending on cell type and the level of oxidative damage, can induce aberrant activation of kinases such as c-Jun N-terminal kinase, p38 MAP kinase, and the extracellular signal-regulated kinase (ERK). If occurring in the neuron, these kinases may contribute to aberrant tau phosphorylation and, if occurring in glial cells, they can contribute to chronic neuroinflammation. Neuroinflammatory responses can lead to aberrant activation cPLA(2)α and AA release, which contribute to the toxic milieu and increased excitotoxicity. For example, Aβ-mediated activation of cPLA(2) α and AA release can be attenuated with NMDA antagonists such as memantine, which is FDA approved for the treatment of AD [92]. In addition, phospholipase A(2) products such as lysophospholipids can perturb membrane phospholipids [92]. Proinflammatory cytokines transcriptionally activate secretory PLA2-IIA in astrocytes via the NF-κB pathway. sPLA2-IIA is an inflammatory protein, which is observed to be associated with neurodegeneration in stroke [93]. NADPH oxidase inhibitors, such as apocynin, and botanical antioxidants, such as resveratrol and epigallocatechin gallate, also inhibited IL-1β-induced sPLA2-IIA mRNA expression, ERK1/2 activation, and translocation of the NADPH oxidase subunit p67 phox from cytosol to membrane fraction [94]. Overall, one could argue that reducing inflammation and AA metabolites should be an important goal of an AD prevention program.
Energy Metabolism (GSK3, Pentose Phosphate Pathway, Glycogenolysis)
Another mechanism known to occur in AD involves stage- and region-dependent defects in energy metabolism. Hypometabolism occurs in the medial temporal lobe (hippocampus, fusiform gyrus, and amygdala) [95]. While ApoE4 patients converting from MCI to AD stage had similar hypometabolism as those not carrying the ApoE4 allele, these subjects showed additional hypometabolism in the frontal cortical areas, such as the anterior cingulate cortex and inferior frontal cortex, consistent with more aggressive pathogenesis in subjects carrying the ApoE4 allele [96].
The metabolic changes are complex, since hypometabolism in vulnerable areas can coexist with hypermetabolism in the inferior temporal/entorhinal cortex as seen in Down's patients [97]. Also, hyperactivation occurs in the prefrontal cortex area associated with auditory–verbal short-term memory, particularly in the left hemisphere [98–100]. Furthermore, during MCI, there are positive correlations between neuroimaging of the amyloid binding PiB and hypermetabolism as measured by FDG PET in the anterior cingulate and precuneus/parietal cortex [101]. The explanations for these patterns remain speculative but could be related to hypermetabolic stages preceding hypometabolism. In animal models, Aβ production appears to positively correlate with brain activity and the sleep–wake cycle [102]. There may be stage-dependent focal loops with Aβ-enhanced excitation increasing Aβ production prior to neurodegeneration and hypometabolic regional stages.
After extensive Aβ deposition has been present in the brain for at least a decade and the onset of cognitive decline is in full swing, there is a good correlation between Aβ and decreased metabolism in brain regions such as the parietal and precuneus cortices. Recently, there have been attempts to model this aspect of AD in animal models where mitochondrial energy defects have been found [103]. Additional factors contributing to energy decline are likely to involve disruption of the pentose phosphate pathway and glycogenolysis [104], while there is some evidence that, in contrast, glycolysis is not affected or even enhanced [105, 106].
Because of in vitro evidence and in vivo tangle-associated elevations in active GSK3 labeling, arguing for its role in tau pathogenesis [107], there has been a common misconception that GSK3 activity is generally elevated in AD. However, total GSK3 protein is actually decreased in AD brain, even inversely and tightly correlating with tangle numbers which may argue against a simple direct role in tangle formation [108]. In fact, compartmentalization is a big issue because inhibited GSK3β, that is, GSK3ser9 (the inactive form), is actually elevated in astrocytes in AD brain where it correlates with glycogen accumulation, an index of metabolic deficits (Frautschy and Hu, unpublished observations). Although GSK3 hyperactivation can mediate the acute effects of Aβ on neurons in vitro and in vivo, the role of GSK3 in AD is far more complex and cell type dependent. Our data argues against GSK3β as an easy therapeutic target [38]. Consistent with recent reports for GSK3β playing a role in memory consolidation and arguing against GSK3β as a good target in AD [107, 109, 110], rats infused with highly specific GSK3 inhibitors in the absence of Aβ showed severe deficits, tau kinase activation (other than GSK3), and impaired memory [38].
GSK3 inhibition in rats increased deposition of glycogen in the brain and similarly in vitro in astrocytes (Frautschy and Hu, unpublished observations). Thus, inhibition of GSK3 in astrocytes may aggravate energy imbalance by stimulating glycogen accumulation. Our unpublished data also shows glycogen accumulation and GSKser9 (inactive form) elevations in AD brain; most noticeably, we have observed upregulation in the hippocampus, while others have found increased GSK3ser9 in the temporal cortex of AD brain [111]. Thus, modulation of aberrant signaling pathways in AD needs to consider possible different alterations in glia versus neurons as well as normal physiological roles. From our perspective, this is another argument for pleiotropic interventions that can indirectly modulate rather than directly inhibit the aberrant activation of tau kinase pathways.
One role of active GSK3 is to normally phosphorylate and inactivate glycogen synthase (GS through its phosphorylation, pGS) and thus limit glycogen synthesis in astrocytes, so elevations in inactive GSK3 should increase glycogen synthesis, and this is exactly what we see, a dramatic increase pGS occurs in astrocytes in Alzheimer's brain compared to normal brain. However, this change was not detected in Western blotting data because immunohistochemical data (of hippocampus) demonstrated that pGS was increased in neurons but decreased in astrocytes (unpublished data). This is consistent with the report that glycogen accumulates in AD brain in the temporal cortex [112]. Using specific antibodies to glycogen, we also observed a robust accumulation of pGS in Alzheimer's brain hippocampus compared to normal age-matched control hippocampus (Frautschy et al., unpublished observations).
Taken together, our unpublished data on the hippocampus and the published data on the temporal cortex suggest that glycogen accumulation is occurring in AD brain. This observation demonstrates that the problem is not simply lack of energy, but an inability to use available stored energy because of defective glycogenolysis, an event similar to Pompe disease in the brain. The precise mechanisms preventing glycogenolysis are unclear since total enzyme levels of glycogen phosphorylase, the first enzyme required for glycogenolysis, were increased but the levels of active glycogen phosphorylase are unknown. Interestingly, another enzyme involved in phosphorylation and activation of glycogen phosphorylase is protein kinase A, which is degraded by calpain and diminished in AD brain [113]. This may be a crucial factor contributing to impaired glycogenolysis. Interestingly, vitamin B6 (pyridoxine), which is an essential cofactor for glycogen phosphorylase, is reduced in Alzheimer's serum [114] and is associated with increased white matter hyperintensities in AD brain [115]. A reduction in phosphatases (e.g., protein phosphatase 1) could lead to hyperphosphorylation of glycogen (amylose) [116], which is resistant to debranching enzymes like α-glucosidase [117]. α-Glucosidase has been reported to be reduced in the temporal cortex of AD brain which may also contribute to increased glycogen and energy deficits [118].
Another important factor limiting glycogen use is the loss of neurotransmitters that stimulate glycogenolysis. For example, denervation of locus coeruleus observed in AD patients and AD models [119]. In addition to norepinephrine, other neurotransmitters are reduced in some brain regions such as serotonin [120], and the combined neurotransmitter deficiency could impede astrocyte glycogenolysis needed for normal brain function.
The last step in glycogenolysis is the production of glucose-6-phosphatase by phosphoglucomutase, which then enters the pentose phosphate pathway. However, glucose-6-phosphate dehydrogenase (G6PD) is needed to maintain NADH but is below 33% in AD [104]. There appears to be no block in glycolysis in AD brain (glucose to pyruvate) since pyruvate is over fivefold elevated in AD CSF [105, 106]. This disproportionate elevation of levels of pyruvate compared to lactate may indicate impaired conversion of pyruvate to lactate for energy, catalyzed by lactate dehydrogenase. This is supported by the fact that NADPH reductase mRNA and protein, which are needed to convert pyruvate to lactate, are also reduced in the hippocampus and/or neocortex of AD brain [121–123]. Defects in the pentose phosphate pathway arising from deficiencies in G6PD will limit NADH needed for conversion to lactate and normal astrocyte function. Lactate is a critical fuel needed to recover synaptic function and to spare glucose as a metabolic fuel.
Promising Pleiotropic Drugs
Docosahexaenoic Acid
DHA is a long-chain 22-carbon omega-3 polyunsaturated fatty acid with six double bonds. This fatty acid is enriched in marine algae, fatty fish, and fish oil. Fish, however, may carry the risk of toxic metal contamination such as mercury [124]. The World Health Organization/Food and Agriculture Organization have determined that the provisional tolerable weekly intake of methylmercury is 80 µg/50 kg body weight per week, while fish oil supplements can have from 9.89 to 123 ng/g oil [125], so recommended guidelines for supplementation with fish oil would be within the tolerable range. For algae DHA, one would receive negligible methylmercury.
DHA is associated with reduced AD risk [126, 127]. Depleting dietary DHA exacerbated AD pathogenesis in a transgenic mouse model, while repleting dietary DHA attenuated AD pathogenesis [128, 129]. Drebrin loss is an early marker of synaptic deficits in MCI, and DHA could remarkably protect against drebrin loss in our AD model mice [128]. Two small trials now show that high-dose fish oil supplements appear to slow progression from very early stage AD [130] or “MCI” [131]. As reported at the International Conference on AD (Vienna, 2009), three larger trials (first the Memory Improvement with DHA Study (MIDAS), funded by Martek, second the Souvenir Trial with the medical food Souvenaid, containing vitamins, uridine, fish oil and choline, funded by Nutricia, Danone [132], and third, the Alzheimer Disease Cooperative Study (ADCS), with DHA, funded by National Institute of Aging) also suggest some efficacy of DHA or fish oil in people, although possibly only beneficial in subjects lacking the ApoE4 allele (see review [133]). DHA appears protective in the epidemiology of dementia and AD, at least in subjects lacking ApoE4, and at least 14 neuroprotective or anti-AD effects of DHA have been reported in preclinical models [134]. Neuroprotective activity found in animals may be relevant to humans where supplementing parental feeding of severe head injury patients with fish oil has improved survival and recovery [135]. DHA's neuroprotective effects in preclinical models include:
-
1.
Anti-inflammatory activity. Unlike conventional NSAIDs that inhibit COX, DHA can competitively reduce CNS levels of AA and, therefore, reduce enzymatic metabolites via COX and LOXs (prostaglandins (PGs) and the leukotrienes like hydroxyeicosatetraenoic acids (HETES), respectively) that are increased by elevated cPLA2. Reduced AA and n-6/n-3 ratios were measured in brains of DHA-fed AD model mice; the n-6/n-3 ratio was down from 0.96 to 0.51 (p < 0.001) [136–138]. Fish oil was shown to reduce AA and cardiovascular risk in arthritis patients consuming NSAIDs, and AA reduction is a well-known NSAID-sparing activity of fish oil in the periphery [139]. This AA metabolite reduction extends also to the brain [140]. Omega-3 is known to reduce NSAID requirements in inflammatory conditions [141], but unlike NSAIDs which may increase cardiovascular risk, omega-3 reduces cardiovascular risk, even in NSAID users [139].
-
2.
Insulin/trophic factor induction of neuroprotective Akt activity is elevated by DHA via increases in phosphatidylserine-regulated membrane docking and Akt activation [142].
-
3.
DHA increases brain-derived neurotrophic factor (BDNF) synthesis [143]. Since BDNF is reduced in AD and is strongly neuroprotective in AD models, this effect may be quite protective. Furthermore, this effect can be enhanced by another safe protective factor, exercise [144]. We have an active research project exploring DHA plus exercise in AD models.
-
4.
Antioxidant. DHA may have direct antioxidant effects in membrane [145] and indirect activity by increasing antioxidant enzymes such as catalase and GSH peroxidase [146].
-
5.
Neuroprotective enzymatic metabolites. DHA metabolites are reported to have anti-apoptotic/anti-inflammatory and other neuroprotective effects, notably from neuroprotectin D1, which is a LOX DHA metabolite that has been reported to exert multiple anti-apoptotic and other anti-AD activities [147].
-
6.
Promotion of neurogenesis and neurite extension. DHA promotes neurite outgrowth in vitro and neurogenesis and improved cognition in vivo [148]. Furthermore, selectively increasing DHA from fat-1 transgene expression has provided additional evidence of these DHA neuroprotective effects in adult mice [149]. DHA stimulation of stem cell neuronal differentiation has been observed in other systems via modulation of cell cycle and basic helix–loop–helix transcription factors [150].
-
7.
While glucose utilization is reduced in AD, DHA increased the expression of a major brain endothelial cell glucose transporter [151].
-
8.
DHA improves the age-impaired coupling of blood flow to glucose utilization in aged monkeys [152]. This may be a factor in both AD and vascular dementia.
-
9.
Neuronal and synaptic membrane fluidity and lipid raft function are believed to decline with aging, while DHA improves synaptic membrane fluidity when it is esterified to membrane phospholipids [153]. Imaging data suggest that this effect can be seen in humans [154]. DHA also enhances lipid raft function [155].
-
10.
DHA increases G protein coupling. While this effect has been established in the retina [156], it may also be important in neuroprotection [157].
-
11.
DHA can activate both peroxisome proliferator-activated receptor (PPAR) and nuclear retinoid X receptor alpha (RXRalpha), involved in the regulation of lipid biosynthesis [158, 159]. This may explain some metabolic and anti-inflammatory effects but the required doses may be slightly supra-physiological and whether RXR activation occurs with physiological DHA dosing remains unclear [160].
-
12.
DHA can protect against oligomer-induced synaptic marker loss in primary neurons [161]. One candidate mechanism involves blocking Aβ-induced phosphorylation of insulin receptor substrate (IRS) by tau kinases. Elevated phospho-IRS is found in AD and AD animal models. This is an important finding because it represents an uncoupling of both insulin and neurotrophic factor signaling to the critical neuroprotective PI3-K/Akt pathway [161].
-
13.
DHA modestly suppressed Aβ production and amyloid accumulation in vitro and in animal models in eight of nine reports [134]. The one study that did not find a reduction in Aβ also reported that dietary DHA failed to increase DHA or reduce AA levels but reduced vascular Aβ [162, 163]. DHA's reduction of Aβ appears to be due to several mechanisms, including altering APP and secretase mobility, reducing the expression of presenilin 1 and gamma secretase activity [137], and induction of increased expression of a late-onset, AD-depleted, anti-amyloidogenic chaperone, SorLa/LR11 [164]. Fish oil has also been reported to increase the expression of transthyretin, which is an Aβ-clearing transport protein [165]. DHA may also increase levels of insulin-degrading enzyme and Aβ clearance by upregulating Akt pathway activity [166].
-
14.
DHA can suppress JNK and GSK3, two important tau kinases that promote tau pathology and neurofibrillary tangles [137, 161].
Taken together, these pleiotropic activities provide multiple pathways for combating AD pathogenesis, first by reducing amyloid (Aβ) production and accumulation, but also multiple ways for suppressing downstream tau kinases and tau pathology, reducing inflammation and oxidative damage, enhancing neuroprotective and neurogenic pathways, and increasing glucose utilization and neuron and synapse function. What is most remarkable is that all of these beneficial activities occur within a range of dosing with proven safety. The one caveat is that DHA is highly susceptible to lipid peroxidation, and a marker for this, F4 isoprostane, is elevated in AD [167, 168]. This implies that AD brain has insufficient antioxidant protection for DHA and provides a strong rationale for combining DHA with protective antioxidants, for example, α-lipoic acid, flavonoids, or vitamin E. There is already limited evidence from one small 12-month clinical trial that combining omega-3 with α-lipoic acid may slow progression in AD [47]. This needs to be confirmed with a larger trial. We have suggested DHA be combined with curcumin, a polyphenolic antioxidant which also has other potent anti-aging, anti-amyloid, and AD protective activities [169]. Combination of curcumin and fish oil in triple transgenic mouse model expressing mutant human APP, PS1, and tau has shown promising results [161].
Curcumin
Curcumin, the yellow pigment found in turmeric root, has been used in medicinal extracts in both Indian traditional Ayurvedic medicine and traditional Chinese and Southeast Asian medicines. Curcumin has pleiotropic effects with antioxidant, anti-inflammatory, anti-carcinogenic [170], and neuroprotective properties [171]. As a medication, curcumin has been used as an anti-inflammatory agent and to promote wound healing in many tissues and disease circumstances. In our recent review, we discuss evidence from many laboratories for several different activities relevant to neuroprotection and AD that include antioxidant, anti-inflammatory, and anti-amyloid activities as well as promoting neurogenesis, heat shock protein synthesis, and limiting the tau and IRS kinase, JNK [171]. These pleiotropic activities of curcumin that are relevant to AD are listed here:
-
1.
Aβ-binding properties: Our group found that curcumin has direct anti-amyloid and anti-Aβ oligomer activity in vitro and in vivo [172–174]. This concept was elegantly demonstrated by Garcia-Alloza et al. [175] who used multi-photon and in vivo imaging to show that animals receiving tail vein injection of curcumin for 1 week had a marked amyloid clearance effect with 30% plaque size reduction in addition to suppression of dystrophic and aberrant neurites [175]. The binding affinity of curcumin for Aβ aggregates is as high as or higher than successful molecular imaging probes such as PiB in FDG PET, with a K i of 0.07 nM for F18-labeled curcumin binding for fibrillar Aβ [176]. In principle, curcumin could also bind other β-pleated sheet structures including prion aggregates [177] as well as synuclein and tau aggregates, but more work needs to be done.
-
2.
Tau-binding properties. Curcumin and related polyphenols have been positive as tau aggregation inhibitors in NIH-funded high-throughput screens [178], but have failed in similar assays where curcumin pyrazole and other derivatives were found to inhibit tau aggregation [179]. These contradictory results may relate to slight differences in methodology, modifying outcome, such as details of tau aggregation kinetics and curcumin solubility, fluorescence, and stability in different systems.
-
3.
Stimulation of phagocytic Aβ clearance: Similar to the amyloid vaccine, curcumin appeared to increase the association of phagocytic cells with plaque structures in a rat AD model [180] and Tg2576 model as well as with plaques in human sections exposed to primary rodent microglia [56]. These effects may derive from modulation of the state of microglial activation through effects on AA metabolites.
-
4.
Anti-inflammatory: Curcumin limits AA substrates and aberrant inflammatory cytokine production. Unlike classical NSAIDs, curcumin does not appear to directly inhibit COX except at high doses (COX-1 [50 µM] or COX-2 [100 µM]) [181]. But like DHA, it has profound effects on limiting multiple AA metabolites. For example, curcumin can reduce COX-2 induction, inhibit 5-LOX (IC50 ∼ 0.7 µM), and limit the release of AA by suppressing the phosphorylation and activation of cPLA2 [182]. Dietary curcumin at 2,000 ppm, a dose with proven long-term safety by the National Toxicology Program [183], was shown to reduce cPLA2 activity in cancer models in vivo [184]. Curcumin's impact on AA metabolism is to counteract AA release and elevation of AA metabolites from COX and LOX [182] that are elevated in AD brain and AD models. 5-LOX and its metabolites are elevated in AD brain [185], and knocking out 5-LOX protects AD Tg models [186], while curcumin inhibits 5-LOX, even at low doses [182]. Unlike COX-2 which is regulated by expression levels, COX-1 activity is primarily regulated by the availability of its substrate AA and curcumin can limit COX-2 expression but more specifically correct the cPLA2 hyperphosphorylation/activation defect leading to elevated AA substrate availability found in AD brain. Curcumin reduces elevations in the cytokine interleukin-1β and TNF-α in the Tg2576 AD mouse model [172]. Aberrant inflammation can increase the expression of Aβ production by increasing BACE [57–60].
-
5.
Anti-ERK and JNK: The mechanism of curcumin's ability to reduce cPLA2 has been investigated in vitro and demonstrated to involve reducing phosphorylation at the same MAPK-sensitive site (ser 505) [182] that is elevated in AD, AD model mice, and Aβ42-stimulated neurons [91]. Consistent with a likely impact on MAPK in brain, low-dose curcumin limits MAPK activation including ERK and MEKK1-JNK in non-neuronal [187] and neuronal PC12 cells in vitro [188].
-
6.
Antioxidant: Unlike ibuprofen, curcumin could effectively protect against oxidative damage in an Alzheimer model [79]. We showed that attenuating inflammation did not attenuate oxidative damage and attenuating oxidative damage with vitamin E did not necessarily reduce inflammation (Frautschy and Hu, unpublished results) or plaque pathogenesis [56]. Similarly, others have reported that indomethacin, the mixed COX-1/COX-2 inhibitor, also failed to suppress oxidative damage by itself, but synergized with the antioxidant vitamin E to reduce both amyloid and oxidative damage [189]. Because of its antioxidant activity, curcumin is typically used as a food preservative and can protect polyunsaturated fatty acids from lipid peroxidation, so it would be predicted to protect and synergize with the highly unsaturated omega-3 fatty acids. Our ongoing studies (unpublished) show that a cocktail containing both DHA and bioavailable curcumin can limit cognitive deficits and tau pathology in human tau mice beginning treatment at 14 months which is well after tau pathology and cognitive deficits had developed [190, 191]. In addition, oxidative damage and increased expression of Aβ production can both be increasing β-secretase BACE [192].
-
7.
Metal chelation. One likely important mechanism of action of curcumin is metal chelation, which requires the dienone bond [193]. Without this bond, for example, when curcumin is reduced to tetrahydrocurcumin, it loses its impact on reducing plaque pathogenesis, but retains its ability to reduce Aβ oligomers [194].
-
8.
Neurogenesis: Curcumin is reported to enhance neurogenesis in two publications [195, 196].
In summary, curcumin is truly pleiotropic and can be used to treat aberrant and chronic inflammation, oxidative damage, amyloid pathology, and tau pathology models.
Curcumin Safety Issues
Curcumin does not share the gastrointestinal bleeds and other toxicity issues of conventional NSAIDs like naproxen and indomethacin and other NSAIDs that limit their use. One explanation for curcumin safety is that curcumin has a dual impact on COX and LOX because dual COX/LOX inhibitors lack the gastrointestinal and cardiovascular safety issues of other NSAIDs that limit their use [197]. Beyond the specific issues of curcumin, pleiotropic interventions may achieve efficacy without side effects by not relying on targeting a single pathway. Like many other age-related chronic diseases, AD does not involve a foreign pathogen but dysregulation of pathways with normal functions. Potential targets include oxygen radicals, COX and AA metabolites, tau, tau kinases, APP, secretases, and even Aβ; all have normal functions and, potently inhibited, these targets can be accepted to result in side effects. Specificity is widely believed to promote safety, which is true when the targets are purely pathogenic, but it does not imply safety when the targets have normal functions and are simply dysregulated in some locations but not others. In contrast, by partially inhibiting multiple branches of the cascade, one may hope to achieve adequate efficacy with fewer side effects.
Bioavailability
Poor bioavailability of curcumin has limited its utility in the clinic [170, 198, 199]. However, we have developed a solid lipid formulation that may help to resolve this problem [194, 200]. The major hurdle for the successful use of curcumin (and related polyphenolics) in human trials has been its poor bioavailability, which stems from poor water solubility, rapid intestinal and first-pass glucuronidation, and instability at cellular pH. For example, no free curcumin was detected in plasma from patients in a clinical trial with 2 or 4 g/day curcumin in AD patients [198]. Similarly, very low levels of unconjugated curcumin were observed in an as yet unpublished University of California, Los Angeles pilot trial in which CSF biomarkers and cognitive endpoints failed to show evidence of efficacy. However, we believe that these obstacles to curcumin use in the clinic can be overcome with new alternative lipidated formulations [194]. In animal model studies, we analyzed plasma and brain levels required for some of the neuroprotective activities and demonstrated that they are achievable with 0.1–0.2 µM plasma levels and associated 1–2 µM brain levels [194]. Lipidated formulations appear to be able to achieve this level and are already in clinical trials for cancer [200] as well as Alzheimer's and other neurodegenerative diseases of aging.
Concluding Remarks
AD is a complicated syndrome of aging with a decades-long prodromal period with accumulation of Aβ and tau pathology accompanied by oxidative damage and inflammation, which develops before an insidious clinical onset. All of these factors can act independently or together to cause neuronal damage and cognitive deficits. By the time clinical deficits emerge, pathology is already proceeding simultaneously at all stages in different brain regions. Interventions need to be as early as possible but targeting single components of the cascade like Aβ should ideally begin early in pathogenesis which is not likely to be feasible with a decades-long prodromal period. Early intervention with safe and inexpensive pleiotropic agents including DHA and curcumin have a strong rationale and in a cocktail may even be effective at later stages due to pleiotropic activities. Problems with energy crisis have not been well addressed clinically, and should be, as they are proximal to cognitive decline.
References
Cole GM, Frautschy SA (2006) Commentary on “Cytoskeletal modulators and pleiotropic strategies for Alzheimer drug discovery.” Pleiotropic approaches to Alzheimer's and other diseases of aging. Alzheimers Dement 2:284–286
Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P, Renbing X, Peck A (1988) Clinical, pathological and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol 23:138–144
Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 45:358–368
Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, Caselli RJ, Kukull WA, McKeel D, Morris JC, Hulette CM, Schmechel D, Reiman EM, Rogers J, Stephan DA (2008) Neuronal gene expression in non-demented individuals with intermediate Alzheimer's Disease neuropathology. Neurobiol Aging 31:549–566
Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ (2010) Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 9:119–128
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356
Morris JC, Storandt M, McKeel DW Jr, Rubin EH, Price JL, Grant EA, Berg L (1996) Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer's disease. Neurology 46:707–719
Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256:184–185
Castellani RJ, Lee HG, Siedlak SL, Nunomura A, Hayashi T, Nakamura M, Zhu X, Perry G, Smith MA (2009) Reexamining Alzheimer's disease: evidence for a protective role for amyloid-β protein precursor and amyloid-β. J Alzheimers Dis 18:447–452
Lee HG, Casadesus G, Zhu X, Takeda A, Perry G, Smith MA (2004) Challenging the amyloid cascade hypothesis: senile plaques and amyloid-β as protective adaptations to Alzheimer disease. Ann N Y Acad Sci 1019:1–4
Ishida A, Furukawa K, Jeffrey NK, Mattson MP (1997) Secreted form of b-amyloid precursor protein shifts the frequency dependency for induction of LTD, and enhances LTP in hippocampal slices. NeuroReport 8:2133–2137
Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O (2008) Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J Neurosci 28:14537–14545
Cole GM, Frautschy SA (2001) Church baptizes Joseph and Perry. Eccentric views absolved. Neurobiol Aging 22:147–150, discussion 161–143
Pimplikar SW (2009) Reassessing the amyloid cascade hypothesis of Alzheimer's disease. Int J Biochem Cell Biol 41:1261–1268
Morihara T, Chu T, Ubeda O, Beech W, Cole GM (2002) Selective inhibition of Aβ42 production by NSAID R-enantiomers. J Neurochem 83:1–4
Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA (2008) Efficacy and safety of tarenflurbil in mild to moderate Alzheimer's disease: a randomised phase II trial. Lancet Neurol 7:483–493
Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH (2009) Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302:2557–2564
Schwab C, Hosokawa M, McGeer PL (2004) Transgenic mice overexpressing amyloid β protein are an incomplete model of Alzheimer disease. Exp Neurol 188:52–64
Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CC, Yang A, Gallagher M, Ashe KH (2006) A specific amyloid-β assembly in the brain impairs memory. Nature 440:352–357
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535–539
Murray CA, Lynch MA (1998) Evidence that increased hippocampal expression of the cytokine interleukin-1 β is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci 18:2974–2981
Hauss-Wegrzyniak B, Vannucchi MG, Wenk GL (2000) Behavioral and ultrastructural changes induced by chronic neuroinflammation in young rats. Brain Res 859:157–166
Hutton M, Lewis J, Dickson D, Yen SH, McGowan E (2001) Analysis of tauopathies with transgenic mice. Trends Mol Med 7:467–470
Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH (2005) Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci 25:10637–10647
Kimura T, Yamashita S, Fukuda T, Park JM, Murayama M, Mizoroki T, Yoshiike Y, Sahara N, Takashima A (2007) Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J 26:5143–5152
Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA (2008) Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372:216–223
Kokjohn TA, Roher AE (2009) Antibody responses, amyloid-β peptide remnants and clinical effects of AN-1792 immunization in patients with AD in an interrupted trial. CNS Neurol Disord Drug Targets 8:88–97
St George-Hyslop PH, Morris JC (2008) Will anti-amyloid therapies work for Alzheimer's disease? Lancet 372:180–182
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 82:239–259
Cummings JL, Cole G (2002) Alzheimer disease. JAMA 287:2335–2338
Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, Mandelkow E, Mandelkow EM (2006) Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem 281:1205–1214
Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 8:663–672
Iqbal K, Liu F, Gong CX, Alonso AD, Grundke-Iqbal I (2009) Mechanisms of tau-induced neurodegeneration. Acta Neuropathol 118:53–69
Bulic B, Pickhardt M, Schmidt B, Mandelkow EM, Waldmann H, Mandelkow E (2009) Development of tau aggregation inhibitors for Alzheimer's disease. Angew Chem Int Ed Engl 48:1740–1752
Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM, Trojanowski JQ (2005) Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci USA 102:227–231
Jope RS, Johnson GV (2004) The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29:95–102
Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH, Yang F, Chen P, Ubeda OJ, Kim PC, Davies P, Ma Q, Cole GM, Frautschy SA (2009) GSK3 inhibitors show benefits in an Alzheimer's disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis 33:193–206
Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, Kowall N (1996) Oxidative damage in Alzheimer's. Nature 382:120–121
Reich EE, Markesbery WR, Roberts LJ 2nd, Swift LL, Morrow JD, Montine TJ (2001) Brain regional quantification of F-ring and D-/E-ring isoprostanes and neuroprostanes in Alzheimer's disease. Am J Pathol 158:293–297
Montine KS, Kim PJ, Olson SJ, Markesbery WR, Montine TJ (1997) 4-Hydroxy-2-nonenal pyrrole adducts in human neurodegenerative disease. J Neuropathol Exp Neurol 56:866–871
Quinn JF, Montine KS, Moore M, Morrow JD, Kaye JA, Montine TJ (2004) Suppression of longitudinal increase in CSF F2-isoprostanes in Alzheimer's disease. J Alzheimers Dis 6:93–97
Behl C, Davis JB, Lesley R, Schubert D (1994) Hydrogen peroxide mediates amyloid β-protein toxicity. Cell 77:817–827
Behl C, Davis J, Cole GM, Schubert D (1992) Vitamin E protects nerve cells from amyloid β-protein toxicity. Biochem Biophys Res Commun 186:944–950
Sung S, Yao Y, Uryu K, Yang H, Lee VM, Trojanowski JQ, Pratico D (2004) Early vitamin E supplementation in young but not aged mice reduces Aβ levels and amyloid deposition in a transgenic model of Alzheimer's disease. FASEB J 18:323–325
Siedlak SL, Casadesus G, Webber KM, Pappolla MA, Atwood CS, Smith MA, Perry G (2009) Chronic antioxidant therapy reduces oxidative stress in a mouse model of Alzheimer's disease. Free Radic Res 43:156–164
Shinto L, Quinn J, Montine T, Baldauf-Wagner S, Oken B, Bourdette D, Kaye J (2008) Omega-3 fatty acids and lipoic acid in Alzheimer's disease. Neurology 70:A393
Zhu X, Castellani RJ, Takeda A, Nunomura A, Atwood CS, Perry G, Smith MA (2001) Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the ‘two hit’ hypothesis. Mech Ageing Dev 123:39–46
Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K (2005) Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer's disease. J Neurosci 25:10180–10187
Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Aggarwal N, Wilson RS, Wilson RS, Scherr PA (2002) Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA 287:33230–33237
Zandi PP, Anthony JC, Khachaturian AS, Stone SV, Gustafson D, Tschanz JT, Norton MC, Welsh-Bohmer KA, Breitner JC (2004) Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch Neurol 61:82–88
Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E et al (1997) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N Engl J Med 336:1216–1222
Lu PH, Edland SD, Teng E, Tingus K, Petersen RC, Cummings JL (2009) Donepezil delays progression to AD in MCI subjects with depressive symptoms. Neurology 72:2115–2121
Wolf G (2007) Estimation of the human daily requirement of vitamin E by turnover kinetics of labeled RRR-alpha-tocopherol. Nutr Rev 65:46–48
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NE, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21:383–421
Cole GM, Morihara T, Lim GP, Yang F, Begum A, Frautschy SA (2004) NSAID and antioxidant prevention of Alzheimer's disease: lessons from in vitro and animal models. Ann N Y Acad Sci 1035:68–84
Morihara T, Teter B, Yang F, Lim GP, Boudinot S, Boudinot FD, Frautschy SA, Cole GM (2005) Ibuprofen suppresses interleukin-1β induction of pro-amyloidogenic alpha1-antichymotrypsin to ameliorate β-amyloid (Aβ) pathology in Alzheimer's models. Neuropsychopharmacology 30:1111–1120
Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT (2003) Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of β-secretase. J Neurosci 23:9796–9804
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O'Banion K, Klockgether T, Van Leuven F, Landreth GE (2005) Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1-42 levels in APPV717I transgenic mice. Brain 128:1442–1453
Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT (2006) Nonsteroidal anti-inflammatory drugs repress β-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci USA 103:443–448
Moore AH, Wu M, Shaftel SS, Graham KA, O'Banion MK (2009) Sustained expression of interleukin-1β in mouse hippocampus impairs spatial memory. Neuroscience 164:1484–1495
Barrientos RM, Frank MG, Hein AM, Higgins EA, Watkins LR, Rudy JW, Maier SF (2009) Time course of hippocampal IL-1 β and memory consolidation impairments in aging rats following peripheral infection. Brain Behav Immun 23:46–54
Li Y, Liu L, Barger SW, Griffin WS (2003) Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci 23:1605–1611
Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ (2000) Complement component C1q modulates the phagocytosis of Aβ by microglia. Exp Neurol 161:127–138
Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E (2002) Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA 99:10837–10842
Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O'Banion MK (2007) Sustained hippocampal IL-1 β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest 117:1595–1604
Mrak RE, Griffin WS (2005) Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging 26:349–354
Frautschy SA, Yang F, Irizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152:307–317
Frautschy S, Cole GM, Baird A (1992) Phagocytosis and deposition of vascular β-amyloid in rat brains injected with Alzheimer β-amyloid. Am J Pathol 140:1389–1399
Wisniewski T, Konietzko U (2008) Amyloid-β immunisation for Alzheimer's disease. Lancet Neurol 7:805–811
Head E, Pop V, Vasilevko V, Hill M, Saing T, Sarsoza F, Nistor M, Christie LA, Milton S, Glabe C, Barrett E, Cribbs D (2008) A two-year study with fibrillar β-amyloid (Aβ) immunization in aged canines: effects on cognitive function and brain Aβ. J Neurosci 28:3555–3566
McGeer EG, McGeer PL (2010) Neuroinflammation in Alzheimer's disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis 19:355–361
Webster S, Lue LF, Brachova L, Tenner AJ, McGeer PL, Terai K, Walker DG, Bradt B, Cooper NR, Rogers J (1997) Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer's disease. Neurobiol Aging 18:415–421
Webster S, Bradt B, Rogers J, Cooper N (1997) Aggregation state-dependent activation of the classical complement pathway by the amyloid b peptide. J Neurochem 69:388–398
Mrak RE, Griffin WS (2005) Potential inflammatory biomarkers in Alzheimer's disease. J Alzheimers Dis 8:369–375
Sapolsky RM (1986) Glucocorticoid toxicity in the hippocampus: reversal by supplementation with brain fuels. J Neurosci 6:2240–2244
Qi D, Rodrigues B (2007) Glucocorticoids produce whole body insulin resistance with changes in cardiac metabolism. Am J Physiol Endocrinol Metab 292:E654–E667
Craft S (2009) The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol 66:300–305
Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao Ashe K, Frautschy SA, Cole GM (2001) Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging 22:983–991
McGeer PL, McGeer EG (2007) NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging 28:639–647
Vlad SC, Miller DR, Kowall NW, Felson DT (2008) Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70:1672–1677
Aisen PS (2008) The inflammatory hypothesis of Alzheimer disease: dead or alive? Alzheimer Dis Assoc Disord 22:4–5
Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, Craft S, Evans D, Green R, Mullan M (2008) Cognitive function over time in the Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol 65:896–905
Laino C (2009) In follow-up analysis of clinical trial, NSAIDs seem to preserve cognitive function in patients with healthy brains. Neurol Today 9:21–22
Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JC, Goodman SN (2004) Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology 23:159–169
Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM (2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci 20(15):5709–5714
Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G (2003) Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci 23:7504–7509
McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A (2008) Ibuprofen reduces Aβ, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res 1207:225–236
Pasqualetti P, Bonomini C, Dal Forno G, Paulon L, Sinforiani E, Marra C, Zanetti O, Rossini PM (2009) A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer's disease. Aging Clin Exp Res 21:102–110
Hoozemans JJ, Rozemuller JM, van Haastert ES, Veerhuis R, Eikelenboom P (2008) Cyclooxygenase-1 and -2 in the different stages of Alzheimer's disease pathology. Curr Pharm Des 14:1419–1427
Sanchez-Mejia RO, Newman JW, Toh S, Yu GQ, Zhou Y, Halabisky B, Cisse M, Scearce-Levie K, Cheng IH, Gan L, Palop JJ, Bonventre JV, Mucke L (2008) Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer's disease. Nat Neurosci 11:1311–1318
Shelat PB, Chalimoniuk M, Wang JH, Strosznajder JB, Lee JC, Sun AY, Simonyi A, Sun GY (2008) Amyloid β peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J Neurochem 106:45–55
Lin TN, Wang Q, Simonyi A, Chen JJ, Cheung WM, He YY, Xu J, Sun AY, Hsu CY, Sun GY (2004) Induction of secretory phospholipase A2 in reactive astrocytes in response to transient focal cerebral ischemia in the rat brain. J Neurochem 90:637–645
Jensen MD, Sheng W, Simonyi A, Johnson GS, Sun AY, Sun GY (2009) Involvement of oxidative pathways in cytokine-induced secretory phospholipase A2-IIA in astrocytes. Neurochem Int 55:362–368
Clerici F, Del Sole A, Chiti A, Maggiore L, Lecchi M, Pomati S, Mosconi L, Lucignani G, Mariani C (2009) Differences in hippocampal metabolism between amnestic and non-amnestic MCI subjects: automated FDG-PET image analysis. Q J Nucl Med Mol Imaging 53:646–657
Mosconi L, Perani D, Sorbi S, Herholz K, Nacmias B, Holthoff V, Salmon E, Baron JC, De Cristofaro MT, Padovani A, Borroni B, Franceschi M, Bracco L, Pupi A (2004) MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET. Neurology 63:2332–2340
Haier RJ, Alkire MT, White NS, Uncapher MR, Head E, Lott IT, Cotman CW (2003) Temporal cortex hypermetabolism in Down syndrome prior to the onset of dementia. Neurology 61:1673–1679
Becker JT, Mintun MA, Aleva K, Wiseman MB, Nichols T, Dekosky ST (1996) Alterations in functional neuroanatomical connectivity in Alzheimer's disease. Positron emission tomography of auditory verbal short-term memory. Ann N Y Acad Sci 777:239–242
Becker JT, Mintun MA, Aleva K, Wiseman MB, Nichols T, DeKosky ST (1996) Compensatory reallocation of brain resources supporting verbal episodic memory in Alzheimer's disease. Neurology 46:692–700
Backman L, Andersson JL, Nyberg L, Winblad B, Nordberg A, Almkvist O (1999) Brain regions associated with episodic retrieval in normal aging and Alzheimer's disease. Neurology 52:1861–1870
Cohen AD, Price JC, Weissfeld LA, James J, Rosario BL, Bi W, Nebes RD, Saxton JA, Snitz BE, Aizenstein HA, Wolk DA, Dekosky ST, Mathis CA, Klunk WE (2009) Basal cerebral metabolism may modulate the cognitive effects of Aβ in mild cognitive impairment: an example of brain reserve. J Neurosci 29:14770–14778
Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM (2009) Amyloid-β dynamics are regulated by orexin and the sleep–wake cycle. Science 326:1005–1007
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 106:14670–14675
Bigl M, Bruckner MK, Arendt T, Bigl V, Eschrich K (1999) Activities of key glycolytic enzymes in the brains of patients with Alzheimer's disease. J Neural Transm 106:499–511
Parnetti L, Reboldi GP, Gallai V (2000) Cerebrospinal fluid pyruvate levels in Alzheimer's disease and vascular dementia. Neurology 54:735–737
Parnetti L, Gaiti A, Brunetti M, Avellini L, Polidori C, Cecchetti R, Palumbo B, Senin U (1995) Increased CSF pyruvate levels as a marker of impaired energy metabolism in Alzheimer's disease. J Am Geriatr Soc 43:316–318
Takashima A (2006) GSK-3 is essential in the pathogenesis of Alzheimer's disease. J Alzheimers Dis 9:309–317
Baum L, Hansen L, Masliah E, Saitoh T (1996) Glycogen synthase kinase 3 alteration in Alzheimer disease is related to neurofibrillary tangle formation. Mol Chem Neuropathol 29:253–261
Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL (2007) LTP inhibits LTD in the hippocampus via regulation of GSK3β. Neuron 53:703–717
Gomez-Sintes R, Hernandez F, Bortolozzi A, Artigas F, Avila J, Zaratin P, Gotteland JP, Lucas JJ (2007) Neuronal apoptosis and reversible motor deficit in dominant-negative GSK-3 conditional transgenic mice. EMBO J 26:2743–2754
Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O'Connor R, O'Neill C (2005) Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem 93:105–117
Mann DM, Sumpter PQ, Davies CA, Yates PO (1987) Glycogen accumulations in the cerebral cortex in Alzheimer's disease. Acta Neuropathol 73:181–184
Liang Z, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2007) Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer disease brain. J Neurochem 103:2462–2470
Mulder C, Scheltens P, Barkhof F, Gundy C, Verstraeten RA, de Leeuw FE (2005) Low vitamin B6 levels are associated with white matter lesions in Alzheimer's disease. J Am Geriatr Soc 53:1073–1074
Mulder C, van der Flier WM, Veerhuis R, Bouwman F, Jakobs C, Verhoeven NM, Barkhof F, Scheltens P, Blankenstein MA (2007) Association between vitamin B6 and white matter hyperintensities in patients with Alzheimer's disease not mediated by homocysteine metabolism. J Am Geriatr Soc 55:956–958
Tagliabracci VS, Turnbull J, Wang W, Girard JM, Zhao X, Skurat AV, Delgado-Escueta AV, Minassian BA, Depaoli-Roach AA, Roach PJ (2007) Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci USA 104:19262–19266
Huang L, Hollingsworth RI, Castellani R, Zipser B (2004) Accumulation of high-molecular-weight amylose in Alzheimer's disease brains. Glycobiology 14:409–416
Cross AJ, Crow TJ, Johnson JA, Dawson JM, Peters TJ (1985) Loss of endoplasmic reticulum-associated enzymes in affected brain regions in Huntington's disease and Alzheimer-type dementia. J Neurol Sci 71:137–143
Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O'Banion MK, Weinberg G, Klockgether T, Feinstein DL (2002) Noradrenergic depletion potentiates β-amyloid-induced cortical inflammation: implications for Alzheimer's disease. J Neurosci 22:2434–2442
Nazarali AJ, Reynolds GP (1992) Monoamine neurotransmitters and their metabolites in brain regions in Alzheimer's disease: a postmortem study. Cell Mol Neurobiol 12:581–587
Nakamura S, Kawamata T, Akiguchi I, Kameyama M, Kimura H (1987) Reduced nicotinamide adenine dinucleotide phosphate diaphorase histochemistry in neocortex and hippocampus in patients with Alzheimer type dementia and aged controls. Rinsho Shinkeigaku 27:1059–1063
Zubenko GS, Moossy J, Claassen D, Martinez AJ, Rao GR (1990) Brain regional analysis of NADH-cytochrome C reductase activity in Alzheimer's disease. J Neuropathol Exp Neurol 49:206–214
Jacobs RW, Farivar N, Butcher LL (1985) Alzheimer dementia and reduced nicotinamide adenine dinucleotide (NADH)-diaphorase activity in senile plaques and the basal forebrain. Neurosci Lett 53:39–44
Tsuchiya A, Hardy J, Burbacher TM, Faustman EM, Marien K (2008) Fish intake guidelines: incorporating n-3 fatty acid intake and contaminant exposure in the Korean and Japanese communities. Am J Clin Nutr 87:1867–1875
Levine KE, Levine MA, Weber FX, Hu Y, Perlmutter J, Grohse PM (2005) Determination of mercury in an assortment of dietary supplements using an inexpensive combustion atomic absorption spectrometry technique. J Automat Meth Manag Chem 2005:211–216
Maclean C, Issa A, Newberry S, Mojica W, Morton S, Garland R, Hilton L, Traina S, Shekelle P (2005) Effects of omega-3 fatty acids on cognitive function with aging, dementia, and neurological diseases. Evid Rep Technol Assess (Summ) (114):1–88
Schaefer EJ, Bongard V, Beiser AS, Lamon-Fava S, Robins SJ, Au R, Tucker KL, Kyle DJ, Wilson PW, Wolf PA (2006) Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: the Framingham Heart Study. Arch Neurol 63:1545–1550
Calon F, Lim GP, Yang F, Morihara T, Teter B, Ubeda O, Rostaing P, Triller A, Salem N Jr, Ashe KH, Frautschy SA, Cole GM (2004) Docosahexaenoic acid protects from dendritic pathology in an Alzheimer's disease mouse model. Neuron 43:633–645
Lim GP, Calon F, Morihara T, Yang F, Teter B, Ubeda O, Salem N Jr, Frautschy SA, Cole GM (2005) A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J Neurosci 25:3032–3040
Freund-Levi Y, Eriksdotter-Jonhagen M, Cederholm T, Basun H, Faxen-Irving G, Garlind A, Vedin I, Vessby B, Wahlund LO, Palmblad J (2006) Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Arch Neurol 63:1402–1408
Chiu CC, Su KP, Cheng TC, Liu HC, Chang CJ, Dewey ME, Stewart R, Huang SY (2008) The effects of omega-3 fatty acids monotherapy in Alzheimer's disease and mild cognitive impairment: a preliminary randomized double-blind placebo-controlled study. Prog Neuropsychopharmacol Biol Psychiatry 32:1538–1544
Scheltens P, Kamphuis PJ, Verhey FR, Olde Rikkert MG, Wurtman RJ, Wilkinson D, Twisk JW, Kurz A (2010) Efficacy of a medical food in mild Alzheimer's disease: a randomized, controlled trial. Alzheimers Dement 6(1–10):e11
Cole GM, Frautschy SA (2010) DHA may prevent age-related dementia. J Nutr 140:869–874
Cole GM, Ma QL, Frautschy SA (2009) Omega-3 fatty acids and dementia. Prostaglandins Leukot Essent Fatty Acids 81:213–221
Heller AR, Rossler S, Litz RJ, Stehr SN, Heller SC, Koch R, Koch T (2006) Omega-3 fatty acids improve the diagnosis-related clinical outcome. Crit Care Med 34:972–979
Calon F, Lim GP, Morihara T, Yang F, Ubeda O, Salem NJ, Frautschy SA, G.M. C (2005) Dietary n-3 polyunsaturated fatty acid depletion activates caspases and decreases NMDA receptors in the brain of a transgenic mouse model of Alzheimer's disease. Eur J NeuroSci 22:617–626
Green KN, Martinez-Coria H, Khashwji H, Hall EB, Yurko-Mauro KA, Ellis L, LaFerla FM (2007) Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-β and tau pathology via a mechanism involving presenilin 1 levels. J Neurosci 27:4385–4395
Oksman M, Iivonen H, Hogyes E, Amtul Z, Penke B, Leenders I, Broersen L, Lutjohann D, Hartmann T, Tanila H (2006) Impact of different saturated fatty acid, polyunsaturated fatty acid and cholesterol containing diets on β-amyloid accumulation in APP/PS1 transgenic mice. Neurobiol Dis 23:563–572
Cleland LG, Caughey GE, James MJ, Proudman SM (2006) Reduction of cardiovascular risk factors with longterm fish oil treatment in early rheumatoid arthritis. J Rheumatol 33:1973–1979
Little SJ, Lynch MA, Manku M, Nicolaou A (2007) Docosahexaenoic acid-induced changes in phospholipids in cortex of young and aged rats: a lipidomic analysis. Prostaglandins Leukot Essent Fatty Acids 77:155–162
Galarraga B, Ho M, Youssef HM, Hill A, McMahon H, Hall C, Ogston S, Nuki G, Belch JJ (2008) Cod liver oil (n-3 fatty acids) as an non-steroidal anti-inflammatory drug sparing agent in rheumatoid arthritis. Rheumatology (Oxford) 47:665–669
Akbar M, Calderon F, Wen Z, Kim HY (2005) Docosahexaenoic acid: a positive modulator of Akt signaling in neuronal survival. Proc Natl Acad Sci USA 102:10858–10863
Rao JS, Ertley RN, Lee HJ, DeMar JC Jr, Arnold JT, Rapoport SI, Bazinet RP (2007) n-3 polyunsaturated fatty acid deprivation in rats decreases frontal cortex BDNF via a p38 MAPK-dependent mechanism. Mol Psychiatry 12:36–46
Wu A, Ying Z, Gomez-Pinilla F (2008) Docosahexaenoic acid dietary supplementation enhances the effects of exercise on synaptic plasticity and cognition. Neuroscience 155:751–759
Yavin E, Brand A, Green P (2002) Docosahexaenoic acid abundance in the brain: a biodevice to combat oxidative stress. Nutr Neurosci 5:149–157
Hossain MS, Hashimoto M, Gamoh S, Masumura S (1999) Antioxidative effects of docosahexaenoic acid in the cerebrum versus cerebellum and brainstem of aged hypercholesterolemic rats. J Neurochem 72:1133–1138
Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG (2005) A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest 115:2774–2783
Innis SM (2007) Dietary (n-3) fatty acids and brain development. J Nutr 137:855–859
He C, Qu X, Cui L, Wang J, Kang JX (2009) Improved spatial learning performance of fat-1 mice is associated with enhanced neurogenesis and neuritogenesis by docosahexaenoic acid. Proc Natl Acad Sci USA 106:11370–11375
Katakura M, Hashimoto M, Shahdat HM, Gamoh S, Okui T, Matsuzaki K, Shido O (2009) Docosahexaenoic acid promotes neuronal differentiation by regulating basic helix-loop-helix transcription factors and cell cycle in neural stem cells. Neuroscience 160:651–660
Pifferi F, Jouin M, Alessandri JM, Haedke U, Roux F, Perriere N, Denis I, Lavialle M, Guesnet P (2007) n-3 Fatty acids modulate brain glucose transport in endothelial cells of the blood-brain barrier. Prostaglandins Leukot Essent Fatty Acids 77:279–286
Tsukada H, Kakiuchi T, Fukumoto D, Nishiyama S, Koga K (2000) Docosahexaenoic acid (DHA) improves the age-related impairment of the coupling mechanism between neuronal activation and functional cerebral blood flow response: a PET study in conscious monkeys. Brain Res 862:180–186
Hashimoto M, Hossain S, Shimada T, Shido O (2006) Docosahexaenoic acid-induced protective effect against impaired learning in amyloid β-infused rats is associated with increased synaptosomal membrane fluidity. Clin Exp Pharmacol Physiol 33:934–939
Hirashima F, Parow AM, Stoll AL, Demopulos CM, Damico KE, Rohan ML, Eskesen JG, Zuo CS, Cohen BM, Renshaw PF (2004) Omega-3 fatty acid treatment and T(2) whole brain relaxation times in bipolar disorder. Am J Psychiatry 161:1922–1924
Wassall SR, Stillwell W (2008) Docosahexaenoic acid domains: the ultimate non-raft membrane domain. Chem Phys Lipids 153:57–63
Litman BJ, Niu SL, Polozova A, Mitchell DC (2001) The role of docosahexaenoic acid containing phospholipids in modulating G protein-coupled signaling pathways: visual transduction. J Mol Neurosci 16:237–242, discussion 279–284
Salem N Jr, Litman B, Kim HY, Gawrisch K (2001) Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids 36:945–959
de Urquiza AM, Liu S, Sjoberg M, Zetterstrom RH, Griffiths W, Sjovall J, Perlmann T (2000) Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science 290:2140–2144
Gani OA, Sylte I (2008) Molecular recognition of docosahexaenoic acid by peroxisome proliferator-activated receptors and retinoid-X receptor alpha. J Mol Graph Model 27:217–224
Calderon F, Kim HY (2007) Role of RXR in neurite outgrowth induced by docosahexaenoic acid. Prostaglandins Leukot Essent Fatty Acids 77:227–232
Ma Q-L, Yang F, Rosario E, Ubeda O, Beech W, Gant D, Chen P, Hudspeth B, Chen C, Zhao Y, Vinters H, Frautschy S, Cole G (2009) Aβ oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via JNK signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci 29:9078–9089
Hooijmans CR, Rutters F, Dederen PJ, Gambarota G, Veltien A, van Groen T, Broersen LM, Lutjohann D, Heerschap A, Tanila H, Kiliaan AJ (2007) Changes in cerebral blood volume and amyloid pathology in aged Alzheimer APP/PS1 mice on a docosahexaenoic acid (DHA) diet or cholesterol enriched Typical Western Diet (TWD). Neurobiol Dis 28:16–29
Hooijmans CR, Van der Zee CE, Dederen PJ, Brouwer KM, Reijmer YD, van Groen T, Broersen LM, Lutjohann D, Heerschap A, Kiliaan AJ (2009) DHA and cholesterol containing diets influence Alzheimer-like pathology, cognition and cerebral vasculature in APPswe/PS1dE9 mice. Neurobiol Dis 33:482–498
Ma QL, Teter B, Ubeda OJ, Morihara T, Dhoot D, Nyby MD, Tuck ML, Frautschy SA, Cole GM (2007) Omega-3 fatty acid docosahexaenoic acid increases SorLA/LR11, a sorting protein with reduced expression in sporadic Alzheimer's disease (AD): relevance to AD prevention. J Neurosci 27:14299–14307
Puskas LG, Kitajka K, Nyakas C, Barcelo-Coblijn G, Farkas T (2003) Short-term administration of omega 3 fatty acids from fish oil results in increased transthyretin transcription in old rat hippocampus. Proc Natl Acad Sci USA 100:1580–1585
Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, Frautschy SA, Cole GM (2004) Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci 24:11120–11126
Nourooz-Zadeh J, Liu EHC, Yhlen B, Anggard EE, Halliwell B (1999) F4-Isoprostanes as specific marker of docosahexaenoic acid peroxidation in Alzheimer's disease. J Neurochem 72:734–740
Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ 2nd, Morrow JD, Montine TJ (2004) Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids 128:117–124
Cole GM, Frautschy SA (2006) Docosahexaenoic acid protects from amyloid and dendritic pathology in an Alzheimer's disease mouse model. Nutr Health 18:249–259
Aggarwal BB, Sundaram C, Malani N, Ichikawa H (2007) Curcumin: the Indian solid gold. Adv Exp Med Biol 595:1–75
Cole GM, Teter B, Frautschy SA (2007) Neuroprotective effects of curcumin. Adv Exp Med Biol 595:197–212
Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM (2001) The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci 21:8370–8377
Cole GM, Yang F, Lim GP, Cummings JL, Masterman DL, Frautschy SA (2003) A rationale for curcuminoids for the prevention or treatment of Alzheimer's disease. Curr Med Chem Immunol Endocr Metab Agents 3:15–25
Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM (2005) Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280:5892–5901
Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ (2007) Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem 102:1095–1104
Ryu EK, Choe YS, Lee KH, Choi Y, Kim BT (2006) Curcumin and dehydrozingerone derivatives: synthesis, radiolabeling, and evaluation for β-amyloid plaque imaging. J Med Chem 49:6111–6119
Caughey B, Raymond LD, Raymond GJ, Maxson L, Silveira J, Baron GS (2003) Inhibition of protease-resistant prion protein accumulation in vitro by curcumin. J Virol 77:5499–5502
Brunden KR, Ballatore C, Crowe A, Smith AB 3rd, Lee VM, Trojanowski JQ (2009) Tau-directed drug discovery for Alzheimer's disease and related tauopathies: a focus on tau assembly inhibitors. Exp Neurol (in press)
Narlawar R, Pickhardt M, Leuchtenberger S, Baumann K, Krause S, Dyrks T, Weggen S, Mandelkow E, Schmidt B (2008) Curcumin-derived pyrazoles and isoxazoles: Swiss army knives or blunt tools for Alzheimer's disease? ChemMedChem 3:165–172
Frautschy SA, Hu W, Kim P, Miller SA, Chu T, Harris-White ME, Cole GM (2001) Phenolic anti-inflammatory antioxidant reversal of Aβ-induced cognitive deficits and neuropathology. Neurobiol Aging 22:993–1005
Huang MT, Lysz T, Ferraro T, Abidi TF, Laskin JD, Conney AH (1991) Inhibitory effects of curcumin on in vitro lipoxygenase and cyclooxygenase activities in mouse epidermis. Cancer Res 51:813–819
Hong J, Bose M, Ju J, Ryu JH, Chen X, Sang S, Lee MJ, Yang CS (2004) Modulation of arachidonic acid metabolism by curcumin and related β-diketone derivatives: effects on cytosolic phospholipase A(2), cyclooxygenases and 5-lipoxygenase. Carcinogenesis 25:1671–1679
Kelloff GJ, Crowell JA, Hawk ET, Steele VE, Lubet RA, Boone CW, Covey JM, Doody LA, Omenn GS, Greenwald P, Hong WK, Parkinson DR, Bagheri D, Baxter GT, Blunden M, Doeltz MK, Eisenhauer KM, Johnson K, Knapp GG, Longfellow DG, Malone WF, Nayfield SG, Seifried HE, Swall LM, Sigman CC (1996) Strategy and planning for chemopreventive drug development: clinical development plan: curcumin. J Cell Biochem Suppl 26:72–85
Rao CV, Rivenson A, Simi B, Reddy BS (1995) Chemoprevention of colon carcinogenesis by dietary curcumin, a naturally occurring plant phenolic compound. Cancer Res 55:259–266
Ikonomovic MD, Abrahamson EE, Uz T, Manev H, Dekosky ST (2008) Increased 5-lipoxygenase immunoreactivity in the hippocampus of patients with Alzheimer's disease. J Histochem Cytochem 56:1065–1073
Firuzi O, Zhuo J, Chinnici CM, Wisniewski T, Pratico D (2008) 5-Lipoxygenase gene disruption reduces amyloid-β pathology in a mouse model of Alzheimer's disease. FASEB J 22:1169–1178
Chen YR, Tan TH (1998) Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene 17:173–178
Gao M, Zhou H, Li X (2009) Curcumin protects PC12 cells from corticosterone-induced cytotoxicity: possible involvement of the ERK1/2 pathway. Basic Clin Pharmacol Toxicol 104:236–240
Yao Y, Chinnici C, Tang H, Trojanowski JQ, Lee VM, Pratico D (2004) Brain inflammation and oxidative stress in a transgenic mouse model of Alzheimer-like brain amyloidosis. J Neuroinflammation 1:21
Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P (2005) Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci 25:5446–5454
Polydoro M, Acker CM, Duff K, Castillo PE, Davies P (2009) Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci 29:10741–10749
Tabaton M (2004) Oxidative stress and β-APP proteolytic processing. Neurobiol Aging 25(S2):S69 (S64-02-03)
Baum L, Ng A (2004) Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer's disease animal models. J Alzheimers Dis 6:367–377, discussion 443–369
Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, Rock CL, Pruitt MA, Yang F, Hudspeth B, Hu S, Faull KF, Teter B, Cole GM, Frautschy SA (2008) Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer's disease. J Pharmacol Exp Ther 326:196–208
Kang SK, Cha SH, Jeon HG (2006) Curcumin-induced histone hypoacetylation enhances caspase-3-dependent glioma cell death and neurogenesis of neural progenitor cells. Stem Cells Dev 15:165–174
Kim SJ, Son TG, Park HR, Park M, Kim MS, Kim HS, Chung HY, Mattson MP, Lee J (2008) Curcumin stimulates proliferation of embryonic neural progenitor cells and neurogenesis in the adult hippocampus. J Biol Chem 283:14497–14505
Rao P, Knaus EE (2008) Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci 11:81s–110s
Baum L, Lam CW, Cheung SK, Kwok T, Lui V, Tsoh J, Lam L, Leung V, Hui E, Ng C, Woo J, Chiu HF, Goggins WB, Zee BC, Cheng KF, Fong CY, Wong A, Mok H, Chow MS, Ho PC, Ip SP, Ho CS, Yu XW, Lai CY, Chan MH, Szeto S, Chan IH, Mok V (2008) Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J Clin Psychopharmacol 28:110–113
Garcea G, Jones DJ, Singh R, Dennison AR, Farmer PB, Sharma RA, Steward WP, Gescher AJ, Berry DP (2004) Detection of curcumin and its metabolites in hepatic tissue and portal blood of patients following oral administration. Br J Cancer 90:1011–1015
Gota VS, Maru GB, Soni TG, Gandhi TR, Kochar N, Agarwal MG (2010) Safety and pharmacokinetics of a solid lipid curcumin particle formulation in osteosarcoma patients and healthy volunteers. J Agric Food Chem 58:2095–2099
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Frautschy, S.A., Cole, G.M. Why Pleiotropic Interventions are Needed for Alzheimer's Disease. Mol Neurobiol 41, 392–409 (2010). https://doi.org/10.1007/s12035-010-8137-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-010-8137-1