
Abstract
The hallmark of adaptive immunity is its ability to recognise a wide range of antigens and technologies that capture this diversity are therefore of substantial interest. New methods have recently been developed that allow the parallel analysis of T cell reactivity against vast numbers of different epitopes in limited biological material. These technologies are based on the joint binding of differentially labelled MHC multimers on the T cell surface, thereby providing each antigen-specific T cell population with a unique multicolour code. This strategy of ‘combinatorial encoding’ enables detection of many (at least 25) different T cell populations per sample and should be of broad value for both T cell epitope identification and immunomonitoring.
Similar content being viewed by others


Introduction
CD8 T cells of the adaptive immune system are essential to combat viral infection, intracellular bacteria, and under certain circumstances, cancer. CD8 T cells utilise their clone-specific T cell receptor (TCR) to recognise cells that display major histocompatibility complex I (MHC-I) molecules that carry a disease-associated (e.g. pathogen-derived) peptide. With the exception of certain immunoprivileged sites, MHC class I molecules are expressed on the surface of all nucleated cells and consequently, CD8 T cells can survey the body for intracellular infections or other diseases that alter the MHC-associated peptide pool. The sequence and hence specificity of the clone-specific TCR that each T cell carries is determined through the rearrangement of the TCRalpha and TCRbeta loci during T cell development. With an estimated number of 1015 possible TCR gene rearrangements (i.e. potential combinations of TCRalpha and TCRbeta chains) [1], the potential TCR repertoire that can be formed is in fact larger than the size of the human T cell compartment (approximately 5 × 1011 cells). Consequently, for most discussions each T cell can in essence be considered unique.
For the monitoring of antigen-specific T cell responses it is obviously of little relevance to follow total T cell frequencies, but rather frequencies of T cells that can recognise a specific antigen should be traced. Over the years, a variety of assay formats have been developed that monitor antigen-specific T cell responses using functional readouts. While these assays are of substantial value to measure specific T cell capacities, these assays fail to detect T cells that lack a particular functional property and consequently cannot be utilised as a generic tool to measure the frequency of antigen-specific T cells.
The detection of antigen-specific T cells independent of their functional activity first became possible with the discovery that such cells can be stained with fluorescently labelled multivalent peptide–MHC (pMHC) complexes. These recombinant pMHC multimers (somewhat mistakenly commonly referred to pMHC tetramers [2]) have over the past years become an essential tool to dissect the antigen-reactivity of T cell populations, as for instance shown by the >2,000 citations to the original paper [3]. However, until recently, MHC multimer-based flow cytometry has been limited to the detection of a single or perhaps two antigen-specific T cell populations per sample, and in particular for clinical samples, this has limited our ability to perform a comprehensive analysis of disease-associated or therapy-induced T cell responses.
The development of a number of new technologies over the past years has brought multiparametric measurement of antigen-specific T cell responses within reach. First, new technologies for pMHC production allow the generation of very large collections of pMHC reagents for T cell analyses. Second, both flow cytometry- and microarray-based high-throughput assays for T cell detection have been developed that can utilise these collections of pMHC reagents. In the following sections we will discuss the techniques that are now available for pMHC-based “high-throughput” analysis of T cell responses. Implementation of these new techniques in immunomonitoring efforts should enable the measurement of T cell reactivity against hundreds to thousands of different pMHC complexes within limited patient material; and consequently broaden our insight into both disease- and therapy-induced T cell recognition.
Detection of antigen-specific T cells by combinatorial encoding
In 2009, two conceptually similar methods for dissecting the antigen-specificity of T cells by use of combinatorial encoding were described [4, 5]. We (Hadrup et al.) developed a technique that uses two-colour combinations of eight different fluorescent labels to enable detection of up to 25 different T cell populations per sample. Simultaneously, Newell et al. published a similar method using all possible combinations of four different fluorescent labels enabling the detection of up to 15 different T cell populations per sample.
The principle of ‘combinatorial encoding’ used in both studies relies on the generation of multi-colour codes on the surface of antigen-specific T cells. Specifically, T cells are incubated with collections of specific pMHC complexes, with each pMHC complex ‘encoded’ by a unique colour combination (Fig. 1). The differently coloured pMHC multimers will assemble on the cell surface of antigen-specific T cells, giving rise to a multi-colour labelled T cell population that can be detected by flow cytometry. By analysing the colour code carried by each individual T cell it is possible to determine its antigen-specificity. Thus, contrary to conventional flow cytometry, in which signal within a single channel is used as a measure of reactivity, specific combinations of colours are used to define antigen-specific T cell populations. When only those colour codes are utilised that are composed of a fixed number of colours (something that we consider preferable, see below) the number of possible unique colour combinations can be described as K(n, r) = n!/r!(n−r)!, with “r” being the number of colours used in each code, and “n” being the number of different fluorescent labels that is used for MHC multimer labelling. We have used two-colour codes (r = 2) of 8 different fluorescent labels (n = 8) that combine to 28 unique dual-colour-codes. Of these 28 possible combinations, 25 were verified to be as efficient in detecting antigen-specific T cells as conventionally labelled PE and APC single-colour tetramers [4].

The principle of combinatorial encoding. Each pMHC multimer is coupled to two different fluorochromes (colours), but each colour can be re-used to encode another pMHC complex but now in combination with a different colour. The resulting pMHC multimers bind to TCR complexes at the T cell surface, resulting in a two-colour labelling of each antigen-specific T cell. Upon analysis by flow cytometry the two-colour code is visualised and can be used to determine the specificity of analysed T cell populations
Combinatorial encoding only becomes a useful approach when the number of fluorescent labels that can be used is sufficiently large (for a two-colour code the break-even point is at three). To extend the number of fluorescent labels used for pMHC multimer labelling beyond the commonly used PE and APC fluorochromes, we have made use of quantum dots (Qdot or QD). The use of quantum dots in flow cytometry as fluorescent labels of both antibodies and MHC multimers has previously been described by the group of M. Roederer [6, 7]. Qdots are semiconductor nanoparticles of cadmium and selenium/tellurium that have an intrinsic fluorescent capacity [8]. They are best excited by light at lower nanometre wavelength (UV to violet nanometre range) and, as the emission spectra of Qdots are very narrow, they are highly suited for multicolour flow cytometry. The currently used Qdots are first coated with a Zinc-shell to improve their optical properties, and then coated with polymer to allow the conjugation of bio-molecules. Streptavidin-coated Qdots with many different emission spectra are available and these can be used for the binding of biotinylated pMHC molecules. We have validated the use of Qdot–MHC multimers for detection of antigen-specific T cells using six different Qdots (QD565, QD585, QD605, QD655, QD705 and QD800) [4]. All QD–MHC complexes detected similar frequencies of antigen-specific T cells as were detected with conventional phycoerythrin (PE) or allophycocyanin (APC) labelled MHC multimers (albeit with varying signal intensity). It should be noted, however, that the efficiency with which different Qdots are detected is highly dependent on the configuration of the flow cytometry system used.
With the combined use of PE, APC and the set of six Qdots, MHC multimer staining can be performed with eight different fluorescent labels and the combination of these fluorochromes into two-colour codes yields the above-mentioned 28 unique colour combinations.
The main advantage of the dissection of antigen-specific T cell responses by combinatorial encoding is that it allows the simultaneous detection of (at present) 25 different T cell populations in the same amount of sample (e.g. peripheral blood) as is normally required for the detection of just one T cell population. By increasing the amount of information on T cell reactivities that can be extracted from a limited amount of patient sample, combinatorial encoding is well-suited for both T cell epitope discovery projects and for the monitoring of immunotherapeutic and vaccination approaches aiming to induce broad T cell responses.
An additional advantage of this first strategy for combinatorial encoding comes from the fact that all antigen-specific T cells are by definition labelled with two and only two MHC multimer colours. Thus, any T cell that is positive in only one or in three or more of the channels used for pMHC detection can be defined as a background event and gated out. Since most background binding in MHC multimer analysis leads to signals in either one or multiple (three or more) channels (Hadrup, unpublished observation), background is reduced approximately tenfold by this gating strategy. In other words, the sensitivity of detecting antigen-specific T cells in a pool of irrelevant T cells is increased about tenfold through the use of this approach for combinatorial encoding, as compared to conventional single-colour MHC multimer staining.
The second combinatorial encoding method for antigen-specific T cell detection, published by Newell et al. [5], does not rely on Qdots, but uses the PE-Cy5 and PE-Cy7 tandem dyes to extend the number of fluorochromes available for MHC multimer labelling. When combined with the APC and PE fluorochromes, this allows the generation of MHC multimers with one of four different labels. Newell et al. use all possible combinations of these four labels to obtain 15 different colour codes. Thus, in contrast to our approach in which all codes have the same number of colours, Newel et al. use both one-, two-, three-, and four-colour encoding. This alternative combinatorial encoding strategy has the advantage of yielding a larger number of colour combinations from a given number of fluorochromes. However, at the same time it prevents the use of the above-described gating strategy, and therefore, likely results in a lower sensitivity.
It is important to note that both combinatorial encoding strategies rely on the assumption that any of the T cell populations that one aims to detect recognises only one of the pMHC complexes present within the staining reaction. If a certain T cell population is reactive with two different pMHC complexes, this T cell population will become labelled with all colours used to encode these two pMHC complexes. When using the gating approach by Hadrup et al., this T cell population would thus be gated out as background events (although with more advanced analysis, a population of cells that does show a consistent signal in three or four channels could still be picked up). When using the approach for combinatorial encoding by Newell et al., such a cross reactive T cell population would not be gated out, but would incorrectly be identified as a T cell population for which the pMHC is encoded by that specific two-, three- or four-colour combination.
How big is this potential issue? For our analyses with pMHC sets of a complexity of 25, we have not noted any instances where such cross-reactivity appears to form a concern. In line with this, even though cross-reactivity of T cells is an intrinsic property of our immune system, required to allow recognition of a vast number of antigens with a limited number of T cells in the human body, the chance of encountering cross-reactivity in a sample of 25 unrelated pMHC complexes appears very limited on theoretical grounds [1]. Specifically, an estimate based on a wide range of murine studies indicates that a given T cell clone can react with approximately 106 different peptides. Thus, with an estimated universe of foreign nonamer peptides of around 1.5 × 1010, the chance that a T cell will cross-react with any randomly chosen peptide is less than one in 104 [1]. Based on this estimate, the chance of finding cross-reactivity within a set of 25 different pMHC complexes is marginal. However, with the development of encoding schemes that allow the simultaneous use of larger peptide libraries this potential problem could become more realistic. More importantly, cross-reactivity could readily become a concern when using combinatorial encoding to detect T cell responses against a set of structurally related peptide antigens, such as different immune escape variants of HIV epitopes [9, 10]. In such cases, single colour-encoding could obviously still be used to generate a footprint of the antigen-reactivity of individual T cells, as has previously been done for influenza A-specific T cells [11].
Detection of antigen-specific T cells by MHC microarrays
Prior to the development of combinatorial encoding, a number of groups have developed MHC microarrays for the high-throughput detection of antigen-specific T cell responses [12–15]. MHC microarrays offer the advantage of spatial resolution of different pMHC complexes when spotted or printed on the microarray surface. Upon incubation, specific T cells can adhere to the counterpart pMHC molecules, resulting in a spatial separation of different antigen-specific T cell populations. MHC microarrays have a very large potential as a high-complexity platform. However, because of significant technical challenges (see below) only few T cell monitoring projects have reported the use of this system [16]. First generation MHC microarrays were generated by direct spotting of the pMHC of interest onto the microarray slide. More recently, linkage of pMHC to the microarray surface via defined DNA probes––an approach known as “nucleic acid cell sorting” (NACS)––has been described [15]. In the latter approach, nucleotide-probes were designed and linked to streptavidin, while the complementary sequence was printed on a conventional DNA microarray slide. MHC multimers of a given specificity were then formed using probe-tagged streptavidin molecules, providing each pMHC complex with a probe that allows binding to a specific site on the microarray. An important advantage of the NACS approach is its high flexibility, since the same DNA-probe array can be coupled with different panels of pMHC probe-tagged streptavidin for use in different T cell analyses.
The major limitations of the current MHC microarrays are their high technical complexity (and consequently low reproducibility, at least across different laboratories) and their relatively low sensitivity. Soen et al. have reported a sensitivity of MHC microarray-based T cell detection of around 0.1% of CD8 T cells [12]. While other groups do report higher sensitivities, these are generally still above the detection limit of MHC multimer-based flow cytometry. The main reason for the relatively low sensitivity of MHC microarrays is likely to be the fact that most systems rely on the passive diffusion of T cells to their target pMHC. Thus, many T cells will simply not reach their target, and be left in solution. As a related issue, if binding is indeed largely restricted to those T cells that happen to land on a spot carrying cognate antigen, the MHC microarray can essentially be considered a miniaturised multiwell assay system, in which T cell reactivity towards many pMHC complexes is analysed in parallel (and in which each pMHC spot forms a “microwell”). In these cases, MHC microarrays may not address the issue of sample size to the same extent as combinatorial encoding, as this requires each T cell to be probed for reactivity against any of the pMHC complexes present within the system.
Of note, a microcirculatory system has been introduced by Deviren et al. and may explain their reported sensitivity of 0.05–0.01% of the analysed cell population, approaching the detection limit of MHC multimer-based flow cytometry [14]. The use of microcirculatory systems is likely to also be essential to allow contact between a given T cell and any of the pMHC complexes present within and thereby preventing the “microwell issue”.
An alternative solution that could improve both the sensitivity and the “microwell issue” is the use of MHC multimer staining in solution with subsequent spatial separation (a strategy only possible on NACS type systems) or an––at present hypothetical––“reverse phase” array format, in which T cells rather than pMHC would be immobilised on the surface and pMHC multimers would be passed over the surface one by one. Such an approach would ensure that all T cells have the potential to encounter relevant pMHC complexes, and serial scanning of the surface by microscopy would allow one to visualise T cells that accumulate a newly added pMHC complex.
If the current issues with MHC microarray-based systems can be solved, MHC microarrays offer substantial potential as a platform for T cell detection. Specifically, the spatial separation of pMHC should support simultaneous detection of hundreds to thousand of different T cell-specificities on a single microarray and the fact that pMHC binding can be combined with the detection of cytokine secretion by co-spotting of cytokine capture antibodies forms a further asset. However, at the current state of technology development, combinatorial encoding does seem the preferable strategy for high-throughput T cell detection, both because of its higher sensitivity and because it provides a solution to the issue of sample size. The different high-throughput methods for T cell epitope analysis developed to date are summarised in Table 1.
Expanding complexity of combinatorial encoding
The number of antigen-specific T cell responses that can be analysed by combinatorial encoding may potentially be expanded in two ways: (1) by inclusion of new fluorochromes and (2) by utilising higher dimension encoding (Fig. 2). Inclusion of additional fluorochromes is an obvious way to combine the two published combinatorial encoding approaches, as the PE-Cy5 and PE-Cy7 tandem dyes used by Newell et al. may potentially be included into the eight-colour scheme used by us––resulting in ten possible fluorochromes for the encoding of pMHC multimers. The addition of two fluorochromes into a two-colour combinatorial encoding scheme would enable the simultaneous detection of up to 45 different T cell-specificities per sample. However, with an increasing number of fluorochromes, higher order-encoding becomes increasingly attractive. Proof of principle has been achieved for three-dimensional combinatorial encoding [4]. With the use of eight different fluorochromes as fluorescent labels for MHC multimers, 56 unique 3-colour (3D) codes can be created, and with inclusion of 2 new fluorescent labels, this number expands to 120 (Fig. 2). Ignoring the obvious challenges with respect to compensation and signal strength for now, two possible issues should be noted. First, a major limitation of moving into 3D combinatorial encoding approaches is the difficulty in data analysis. A select number of flow cytometry software packages, such as FlowJo, support visualisation in 3D. However, these new software additions are at present not suited for high-throughput analyses. Attempts are currently made to design new flow cytometry software that would allow the automation of flow cytometry analysis, by defining clusters of cells in predefined colour combinations. This approach would minimise subjective ‘gating’ of cells and would be well-suited for higher dimension (3D/4D) analyses (personal communication, Dr. C. Chan, Duke University). Second, when moving to analyses with an increasing number of pMHC multimers in one sample, generation of the mixtures of pMHC complexes may become a challenge. High-throughput generation of pMHC molecules by UV-induced peptide exchange (see below) has a concentration limit of about 200–400 μg/mL. When using each individual pMHC at a final concentration of around 2 μg/mL this will allow for the generation of pMHC pools with a complexity of 100–200. Concentration of pMHC multimer mixes is possible, but often associated with considerable losses. Thus, with the current approaches for high-throughput T cell analyses by combinatorial encoding, a complexity of 100–200 different pMHC multimers per sample is likely to form a practical limit.

The number of possible colour combinations. The graph depicts the number of possible colour combinations (y-axes) that can be made based on one-, two-, or three-colour codes, with a given number of fluorescent labels available (x-axes)
“High-throughput” pMHC production
The development of combinatorial encoding for the parallel detection of antigen-specific T cells forms the most recent addition to a number of methods that aim to make MHC-based analyses suitable for high-throughput screening. Most importantly, the generation of vast numbers of different pMHC complexes necessary for multiplexed T cell analyses has been made possible by the development of UV-induced peptide exchange technology [17, 18]. This technology relies on the design of a UV-sensitive (and therefore ‘conditional’) MHC ligand. Upon UV exposure, this ligand is cleaved and the resulting peptide fragments will quickly disassociate from the peptide binding groove. This results in the generation of a peptide-free (and hence very unstable) MHC molecule that can be rescued by the binding of a new peptide ligand. This peptide ligand can be any ligand of interest, with the obvious caveat that MHC rescue will depend on the MHC affinity of the new ligand [19, 20]. By use of this technology, parallel production of vast numbers of pMHC complexes can be accomplished in a few hours and is feasible in both 96- and 384-well formats. Structural studies have demonstrated that the pMHC complex that is formed upon ligand exchange is identical to that obtained by classical refolding [21]. Furthermore, because ligand exchange takes place under physiological conditions, MHC reagents can be prepared with peptide ligands that display only a moderate affinity for MHC, such as the unmodified MART-I HLA-A2 epitope [17].
An alternative strategy for generating pMHC complexes was recently described by Leisner et al. [22] and relies on the high folding efficiency of MHC class I heavy chains that carry pre-oxidised disulphide bonds. Upon folding of these pre-oxidised (and already biotinylated) heavy chains with β2-microglobulin and peptide, the resulting pMHC complexes can be used for pMHC multimer production without further purification or concentration. At present, this method has not been utilised in larger T cell monitoring studies and such studies will be important to determine its broader value.
In addition to the use of UV-induced peptide exchange for the generation of pMHC reagents for T cell staining, this ligand exchange technology can also be used to measure the affinity of putative MHC-ligands in high-throughput assays. “MHC rescue” after peptide exchange can readily be measured by an MHC-ELISA that measures the concentration of correctly folded MHC that remains after UV exposure. This MHC-ELISA gives a rough estimate of peptide affinity, sufficient for most T cell epitope discovery projects and can be performed in both 96- and 384-well formats, allowing the screening of hundreds or thousands of candidate peptides. Furthermore, a more accurate measure of the MHC affinity of a given peptide can be obtained by peptide exchange-based fluorescence polarisation assays, in which competition of an experimental ligand with a fluorescently labelled high-affinity ligand is quantified [18].
A commercially available platform known as “iTopia™ Epitope Discovery System” (Beckman Coulter) offers an alternative “high-throughput” strategy for the analysis of peptide HLA affinity. The iTopia™ platform relies on denaturation of a reference pMHC complex followed by reconstitution in the presence of the peptide of interest, and the efficiency of this reconstitution depends on the affinity of the peptide for the HLA molecule in question. A number of T cell epitope discovery projects have made use of this system [23–26].
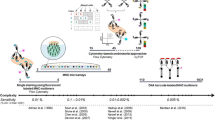
A workflow for ‘high-throughput’ T cell epitope mapping
The majority of current T cell epitope discovery projects are based on ‘reverse immunology’. Starting off with the selection of target genes of interest (e.g. a viral genome), potential MHC-ligands within the encoded protein sequence are identified and subsequently, it is established whether the identified MHC-ligands are recognised by T cells (Fig. 3).

Schematic overview of ‘high-throughput’ T cell epitope mapping. The combination of three MHC-based technologies. a the generation of pMHC complexes by UV-induced exchange of a conditional MHC ligand. b Peptide-MHC affinity screening by MHC-ELISA. c T cell detection by combinatorial encoding, together forms a high-throughput platform for T cell epitope mapping by ‘reverse immunology’
Web-based prediction databases are available to assist the selection of potential MHC–ligands [27, 28]. However, depending on the cut-off chosen, these databases either miss a substantial number of MHC–ligands or provide many more potential ligands than actually do bind to the relevant MHC allele product. The combination of algorithm-based prediction with a quick screening for MHC affinity provides a solution to this issue. Specifically, using a low cut-off in epitope prediction will ensure a low percentage of false-negatives but at the cost of having a high number of false-positives. When this starting collection is subsequently analysed for MHC binding using one of the above-described binding assays, this can reduce the number of candidate peptides to 30–50%, providing an important focus of the peptide library. As the success rate of T cell epitope mapping projects critically depends on the quality of the selected set of putative epitopes used for T cell screening, this combination of in silico prediction and subsequent in vitro screening for peptide-MHC affinity represents a useful strategy to focus the downstream effort on those peptides that are most likely to represent true T cell epitopes.
The combined use of in silico prediction, MHC peptide exchange-based measurement of actual HLA affinity, and detection of T cell responses by combinatorial encoding, has successfully been used for T epitope discovery. In a pilot project we have used this combination of strategies to identify new HLA-A3-restricted T cell epitopes from melanoma-associated proteins. Using a low cut-off algorithm-based prediction, we selected 256 potential HLA-A3 ligands from four melanoma-associated proteins. Using affinity measurements, a set of 22 peptides with the highest HLA-A3 affinity was subsequently identified, and this set was used to screen for T cell reactivity in peripheral blood from melanoma patients by combinatorial encoding. In this specific case, a pre-enrichment step was included, using PE-labelled MHC tetramers to label antigen-specific T cells and isolate them with the aid of anti-PE coated magnetic beads, to increase the sensitivity of detection. Using this strategy, T cell reactivity against eight peptide ligands was detected, of which three were previously described HLA-A3 T cell epitopes and five were potentially new T cell epitopes. While it remains to be established that these new T cell epitopes are actually presented on the surface of tumour cells, this pilot screening project demonstrates the feasibility of high-throughput MHC-based screening projects for the identification of novel T cell epitopes.
Conclusion
‘High-throughput’ approaches for T cell epitope discovery and monitoring, have the potential to significantly improve our understanding of disease- and therapy-induced T cell reactivity. The human body holds approximately 2 × 1011 different T cell clones, and T cell recognition of pathogen-derived or cancer-associated epitopes is therefore likely to be complex [1]. A glimpse of our lack of understanding of the breadth of T cell reactivity becomes apparent when analysing the MHC restriction of the epitopes identified to date. For both infectious disease- and cancer-associated epitopes, a very strong bias towards restriction by the dominant HLA alleles in the European and North-American populations is apparent. As an example, a compilation of the melanoma-associated epitope set described to date for the HLA alleles A1, A2, A3, A11, A24, B7, B57 that we performed for a planned immunomonitoring project revealed that about 70% of these epitopes were restricted by a single HLA allele, HLA-A2, which is the most prevalent HLA allele in the Western European population (Hadrup SR, unpublished data). Thus, there is a clear need for the identification of T cell epitopes restricted by other HLA-types, as 60% of the Western European population does not express HLA-A2, and this number is even substantially higher for all other populations.
A broader knowledge of the disease-associated epitope repertoire is likely to be particularly valuable when monitoring T cell responses induced in a non-HLA restricted fashion. To provide two specific examples, treatment of patients with metastatic melanoma with ex vivo expanded tumour-infiltrating lymphocytes has shown highly encouraging data in a single centre study [29, 30] and anti-CTLA4 is currently in phase III trials for the same patient group (ClinicalTrials.org: NCT00636168, Bristol-Myers Squibb and NCT00257205/NCT00584493, Pfizer). While in both cases clinical effects are likely to be mediated at least in part by CD8 T cells, at present we have very little information on the antigen reactivities involved. With the development of ‘high-throughput’ T cell epitope mapping approaches we now have the tools to obtain a more comprehensive view of antigen-specific T cell recognition that forms the basis of these emerging therapies.
References
Mason D (1998) A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today 19:395–404
Bakker AH, Schumacher TN (2005) MHC multimer technology: current status and future prospects. Curr Opin Immunol 17:428–433
Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM (1996) Phenotypic analysis of antigen-specific T lymphocytes. Science 274:94–96
Hadrup SR, Bakker AH, Shu CJ, Andersen RS, van Veluw VJ, Hombrink P, Castermans E, Thor SP, Blank C, Haanen JB, Heemskerk MH, Schumacher TN (2009) Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods 6:520–526
Newell EW, Klein LO, Yu W, Davis MM (2009) Simultaneous detection of many T-cell specificities using combinatorial tetramer staining. Nat Methods 6:497–499
Perfetto SP, Chattopadhyay PK, Roederer M (2004) Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol 4:648–655
Chattopadhyay PK, Price DA, Harper TF, Betts MR, Yu J, Gostick E, Perfetto SP, Goepfert P, Koup RA, De Rosa SC, Bruchez MP, Roederer M (2006) Quantum dot semiconductor nanocrystals for immunophenotyping by polychromatic flow cytometry. Nat Med 12:972–977
Hotz CZ (2005) Applications of quantum dots in biology: an overview. Methods Mol Biol 303:1–17
Varela-Rohena A, Molloy PE, Dunn SM, Li Y, Suhoski MM, Carroll RG, Milicic A, Mahon T, Sutton DH, Laugel B, Moysey R, Cameron BJ, Vuidepot A, Purbhoo MA, Cole DK, Phillips RE, June CH, Jakobsen BK, Sewell AK, Riley JL (2008) Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat Med 14:1390–1395
Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EL, Salazar MG, Weinhold KJ, Moore S, Letvin N, Haynes BF, Cohen MS, Hraber P, Bhattacharya T, Borrow P, Perelson AS, Hahn BH, Shaw GM, Korber BT, McMichael AJ (2009) The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med 206:1253–1272
Haanen JB, Wolkers MC, Kruisbeek AM, Schumacher TN (1999) Selective expansion of cross-reactive CD8+ memory T cells by viral variants. J Exp Med 190:1319–1328
Soen Y, Chen DS, Kraft DL, Davis MM, Brown PO (2003) Detection and characterization of cellular immune responses using peptide–MHC microarrays. PLoS Biol 1:429–438
Stone JD, Demkowicz WE Jr, Stern LJ (2005) HLA-restricted epitope identification and detection of functional T cell responses by using MHC–peptide and costimulatory microarrays. Proc Natl Acad Sci USA 102:3744–3749
Deviren G, Gupta K, Paulaitis ME, Schneck JP (2007) Detection of antigen-specific T cells on p/MHC microarrays. J Mol Recognit 20:32–38
Kwong GA, Radu CG, Hwang K, Shu CJ, Ma C, Koya RC, Comin-Anduix B, Hadrup SR, Bailey RC, Witte ON, Schumacher TN, Ribas A, Heath JR (2009) Modular nucleic acid assembled p/MHC microarrays for multiplexed sorting of antigen-specific T cells. J Am Chem Soc 131:9695–9703
Chen DS, Soen Y, Stuge TB, Lee PP, Weber JS, Brown PO, Davis MM (2005) Marked differences in human melanoma antigen-specific T cell responsiveness after vaccination using a functional microarray. PLoS Med 2:1018–1030
Toebes M, Coccoris M, Bins AD, Rodenko B, Gomez R, Nieuwkoop NJ, van de Kasteele W, Rimmelzwaan G, Haanen JB, Schumacher TN (2006) Design and use of conditional MHC class I ligands. Nat Med 12:246–251
Bakker AH, Hoppes R, Linnemann C, Toebes M, Rodenko B, Berkers CR, Hadrup SR, van Esch WJ, Heemskerk MH, Ovaa H, Schumacher TN (2008) Conditional MHC class I ligands and peptide exchange technology for the human MHC gene products HLA-A1, -A3, -A11 and -B7. Proc Natl Acad Sci USA 105:3825–3830
Rodenko B, Toebes M, Hadrup SR, van Esch WJ, Molenaar AM, Schumacher TN, Ovaa H (2006) Generation of peptide–MHC class I complexes through UV-mediated ligand exchange. Nat Protoc 1:1120–1132
Hadrup SR, Toebes M, Rodenko B, Bakker AH, Egan DA, Ovaa H, Schumacher TN (2009) High-throughput T-cell epitope discovery through MHC peptide exchange. Methods Mol Biol 524:383–405
Celie PH, Toebes M, Rodenko B, Ovaa H, Perrakis A, Schumacher TN (2009) UV-induced ligand exchange in MHC class I protein crystals. J Am Chem Soc 131:12298–12304
Leisner C, Loeth N, Lamberth K, Justesen S, Sylvester-Hvid C, Schmidt EG, Claesson M, Buus S, Stryhn A (2008) One-pot, mix-and-read peptide–MHC tetramers. PLoS One 3:e1678
Semeniuk CA, Capina RE, Mendoza MG, Kimani J, Ball TB, Luo M, Plummer FA (2009) Identification and characterization of HLA-A*0301 epitopes in HIV-1 gag proteins using a novel approach. J Immunol Methods 352:118–125
Wulf M, Hoehn P, Trinder P (2009) Identification of human MHC class I binding peptides using the iTOPIA-epitope discovery system. Methods Mol Biol 524:361–367
Shingler WH, Chikoti P, Kingsman SM, Harrop R (2008) Identification and functional validation of MHC class I epitopes in the tumor-associated antigen 5T4. Int Immunol 20:1057–1066
Bachinsky MM, Guillen DE, Patel SR, Singleton J, Chen C, Soltis DA, Tussey LG (2005) Mapping and binding analysis of peptides derived from the tumor-associated antigen survivin for eight HLA alleles. Cancer Immun 22:5–6
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S (1999) SYFPEITHI: database for MHC-ligands and peptide motifs. Immunogenetics 50:213–219
Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O, Buus S (2007) NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One 2:e796
Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA (2005) Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 23:2346–2357
Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, Wunderlich J, Restifo NP, Thomasian A, Downey SG, Smith FO, Klapper J, Morton K, Laurencot C, White DE, Rosenberg SA (2008) Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol 26:5233–5239
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is a Focussed Research Review based on a presentation given at the Seventh Annual Meeting of the Association for Immunotherapy of Cancer (CIMT), held in Mainz, Germany, 3–5 June 2009.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hadrup, S.R., Schumacher, T.N. MHC-based detection of antigen-specific CD8+ T cell responses. Cancer Immunol Immunother 59, 1425–1433 (2010). https://doi.org/10.1007/s00262-010-0824-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-010-0824-2