
Abstract
Progression of colon cancer from microadenoma to macroscopic tumors is coupled to augmentation of canonical Wnt signaling. We recently reported that tumor associated macrophages, through interleukin 1β (IL-1β) dependent phosphorylation of GSK3β, promote Wnt signaling in colon cancer cells, demonstrating that proinflammatory cytokines can enhance TCF4/β-catenin transcriptional activity in tumor cells. Here we investigated the pathway whereby IL-1β inactivates GSK3β and promotes Wnt signaling in colon cancer cells. We showed that normal human monocytes, THP1 macrophages and IL-1 failed to induce Wnt signaling in tumor cells expressing dominant negative IκB (dnIκB), demonstrating that macrophages and IL-1 activate Wnt signaling in a NF-κB-dependent manner. NF-κB activity was required for macrophages and IL-1 to activate PDK1 and AKT in tumor cells and thereby inhibit GSK3β activity. Consistently, dominant negative AKT (dnAKT), or pharmacological inhibition of AKT in tumor cells, prevented macrophage/IL-1 mediated phosphorylation of GSK3β, activation of Wnt signaling, and induction of c-jun and c-myc, confirming that macrophages and IL-1 promote Wnt signaling in an AKT dependent manner. Finally, we showed IL-1 and macrophages failed to promote growth of colon cancer cells with impaired NF-κB or AKT signaling, confirming the requirement for NF-κB and AKT activation for the protumorigenic activity of tumor associated macrophages. Thus, we showed that IL-1 and tumor associated macrophages activate NF-κB-dependent PDK1/AKT signaling in tumor cells, and thereby inactivate GSK3β, enhance Wnt signaling and promote growth of colon cancer cells, establishing a novel molecular link between inflammation and tumor growth.
Similar content being viewed by others

Introduction
Tumor associated macrophages (TAMs) are derived from circulating monocytes which, upon recruitment to the tumor microenvironment, polarize and acquire several properties of M2 macrophages [1, 2]. The tumor microenvironment therefore “educates” macrophages to orchestrate conditions that support tumor progression and promote metastasis and angiogenesis [3].
We recently demonstrated that colon cancer cells stimulate normal human monocytes and THP1 macrophages to release IL-1β, and showed that IL-1β is sufficient to induce canonical Wnt signaling and to promote growth of colon cancer cells through inactivation of GSK3β in the epithelial cells, establishing a previously unknown link among inflammation, IL-1β, Wnt signaling and growth of colon cancer cells (Kaler et al, in press). Macrophages/IL-1β induced Wnt signaling in a panel of colon cancer cell lines, including HCT116, Hke-3, SW480 and RKO cells (not shown). It remains to be determined whether macrophages/IL-1β regulate the expression and the activity of Wnt ligands, Wnt receptors or Wnt inhibitors, however we showed that macrophages provoked phosphorylation of GSK3β, stabilized β-catenin and enhanced TCF4-dependent gene activation and the expression of Wnt target genes in tumor cells. In this regard, β-catenin translocation is often detected at the invasive front between the tumor and surrounding tissue [4, 5], consistent with the hypothesis that surrounding tissue at the invasion front provides soluble factors that promote nuclear translocation of β-catenin in tumor cells and thus drive tumor progression. Although increased density of TAMs (tumor associated macrophages) is associated with poor prognosis in breast, prostate, bladder and cervical cancer [6–11], there are contrasting reports regarding the prognostic significance of macrophage infiltration in colon cancer [12–14]. Our findings support a protumorigenic role of tumor associated macrophages in colon cancer, and suggest that they promote tumor growth, at least in part, through secretion of IL-1β.
IL-1β is a proinflammatory cytokine that plays an important role in inflammation, regulates the immune response and is abundant at tumor sites [15]. Chemically induced tumor formation was shown to be significantly delayed in IL-1β deficient mice and IL-1Ra−/− mice, which have excessive levels of IL-1β, display rapid tumor development and high tumor frequency [15–17]. Consistently, the growth of B16 cell xenografts, and their spread to lung and liver were reduced in IL-1β−/− mice [17, 18] and reduced hepatic metastases were seen in mice that lack IL-1β converting enzyme (ICE, caspase 1) [18], confirming that IL-1β can regulate both initiation and the progression of cancer. The IL-1 signaling pathway is well characterized and it has been shown that IL-1 recruits Myd88 to the IL-1 receptor, which connects the receptor with a downstream kinase, IRAK [19]. A dominant negative Myd88 inhibits IL-1 induced activation of NF-κB, a major signaling pathway utilized by IL-1 [19]. Importantly, deficiency in Myd88 has been shown to significantly attenuate intestinal polyposis in Apcmin/+ mice and to increase their survival [20], demonstrating that Myd88 dependent signaling critically contributes to intestinal tumorigenesis. Several inflammatory mediators are increased in ApcMin/+ polyps, including IL-1 [20], suggesting that the decreased tumor number in the ApcMin/+/Myd88−/−compound mouse may be due to deficient signaling by IL-1.
In this study we investigated the pathway whereby macrophages/IL-1 inactivate GSK3β, promote Wnt signaling and enhance growth of colon cancer cells. NF-κB has been shown to regulate the survival of tumor cells and to link inflammation and tumor progression [21–23]. We showed that macrophages, like IL-1, activate NF-κB signaling in colon cancer cells, leading to activation of the AKT pathway. PKB/AKT is a kinase that is activated by recruitment to the plasma membrane through phosphorylation on Thr308 by PDK1 and on Ser473 by PDK2 [24, 25]. It has a crucial role in promoting cell survival through phosphorylation of Bad [26], caspase-9 [27], FKHR [28] and IKKα [29]. Another important downstream target of AKT is GSK3β [30], a kinase with a crucial role in Wnt signaling. The pool of GSK3β that participates in Wnt signaling is present in a multiprotein complex that includes axin, β-catenin and APC [31, 32]. In the absence of Wnt signaling, GSK3β phoshorylates axin, β-catenin and APC, which targets β-catenin for ubiquitin mediated degradation. Wnt signaling results in inactivation of GSK3β, which leads to dephosphorylation of axin, APC and β-catenin [33]. Unphosphorylated β-catenin is stabilized and translocates to the nucleus, where it binds to members of the TCF family of transcription factors, and finally stimulates the expression of target genes such as c-myc, c-jun, CD44 and cox-2 [34].
In this study we established that IL-1 and tumor associated macrophages inactivate GSK3β and promote Wnt signaling in tumor cells through NF-κB dependent activation of PDK1 and AKT. Our data therefore suggest that inhibitors of the NF-κB and PI3K/AKT pathways, which are in development as chemotherapeutic agents, may not only work by inhibiting proliferation and promoting apoptosis of tumor cells, but may also interrupt the crosstalk between the tumor cells and stroma and thereby stall tumor progression.
Experimental Procedures
Cell lines and Coculture Experiments
The HCT116 colorectal carcinoma cell line and its clonal derivative, Hke-3 cells, which lack the mutant k-Ras allele [35] were cultured under standard conditions in minimum essential medium supplemented with 10% fetal calf serum and antibiotics. The human monocytic cell line, THP1, was cultured in RPMI medium. Normal human monocytes, >90% CD14 and CD11c positive and less than 1% anti T cell receptor positive, were purchased from Astarte Biologics (Redmond, WA). Tumor cells and monocytes/macrophages were co-cultured separated by transwell inserts of a polycarbonate membrane with 0.4 μM pore size, thus precluding direct cell-cell contact, but permitting the exchange of soluble factors (Corning Incorporated, Lowell, MA).
Transient Transfections and Reporter Gene Assay
HCT116 and HKe-3 cells were grown in 12-well plates and were transiently transfected with 0.5 µg of luciferase reporter plasmids per well using the calcium phosphate method (Profection mammalian Transfection system, Promega, Madison, WI). Transfection efficiency was normalized by co-transfection with pTK-Renilla, and luciferase activity was determined according to the vendor’s protocol (Dual Luciferase reporter assay, Promega, Madison, WI). Dominant negative IκBα was expressed from a plasmid that codes for IκBα with serines 32 and 36 mutated to alanine, which confers resistance to stimulus induced degradation [36]. Plasmids expressing constitutively active AKT, (HA-mdelta (4-129) PH-AKT), and dominant negative AKT (HA-AKT-K179M) were provided by Richard Roth [26, 37].
IL-1β and STAT1 were silenced in THP1 macrophages by transient transfection with 20 nM of siRNAs specific for IL-1β or STAT1 (Dharmacon, Lafayette, CO) using Lipofectamine LTX (Invitrogen, Carlsbad, CA) as we described earlier (Kaler et al, in press, [38]).
Clonogenic Assay
To asses the clonogenic potential of HCT116 and Hke-3 cells and the effect of macrophage-derived factors on their clonogenic potential, tumor cells were seeded at a density of 200 or 400 cells per well of a six well plate and were cultured with THP1 cells or were treated with IL-1 for 4 days. Cells were then washed and grown in complete media for another 3 days. Colonies were washed with PBS, fixed and stained with 6% glutaraldehyde and 0.5% crystal violet for 30 min at room temperature. Colonies were counted and their average volume determined using Total Lab 1.1 software (Nonlinear Dynamics, Durham, NC, USA).
Immunoblotting
Proteins were fractionated by 10% SDS-PAGE and transferred onto a nitrocellulose membrane. Membranes were blocked with 5% nonfat dry milk in TBS containing 0.1% Tween 20 and then incubated with antibodies specific to pAKT (Ser473), pAKT (Thr308), total AKT, pPDK1, p-cRaf, pGSK3β, active β-catenin, phospho-c-Myc (Thr58/Ser62) (Cell Signaling Technology, Inc. Danvers, MA), β-actin (Sigma Aldrich, St. Louis, MO), c-Myc, c-Jun (Santa Cruz Biotechnology Inc., Santa Cruz, CA); HA (Roche Applied Science, Indianapolis, IN); and IκBα (New England Biolabs, Ipswich, MA). Bound antibodies were detected using peroxidase-coupled secondary antibodies, followed by an enhanced chemiluminescent detection system (Amersham ECL™ western blotting detection kit, Amersham Biosciences, Piscataway, NJ).
Results
Macrophages/IL-1β Induce Wnt Signaling in a NF-κB Dependent Manner
We recently demonstrated that IL-1β induces Wnt signaling in colon cancer cells, a novel signaling pathway for this cytokine (Kaler et al, in press). We showed that IL-1 failed to induce the expression of c-jun and c-myc in cells transfected with dnTCF4 (not shown), confirming that the expression of at least some IL-1 target genes requires intact Wnt signaling.
We recently showed that colon cancer cells stimulate normal peripheral blood monocytes and THP1 macrophages to release IL-1β (Kaler et al, in press). Consistent with the IL-1 release, THP1 macrophages increased NF-κB transcriptional activity in HCT116 cells (Fig. 1A), and normal peripheral blood monocytes and THP1 cells induced degradation of IκBα in both HCT116 and Hke-3 colon cancer cell lines (Fig. 1B).

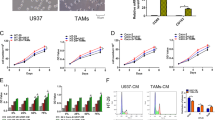
THP1 macrophages induce NF-κB signaling in HCT116 cells. a HCT116 cells were transiently transfected with the NF-κB reporter gene in the absence or the presence of dnTCF4 as indicated, and were cultured alone or together with THP1 macrophages for 24 h. b HCT116 and Hke-3 cells were co-cultured with normal peripheral blood monocytes, THP1 macrophages or were treated with IL-1 (5 ng/ml) as indicated and the levels of IκBα was determined by immunoblotting, c and d HCT116 cells were transfected with the NF-κB reporter plasmid (C) or the TOP-FLASH reporter (D) together with an empty plasmid (neo) or dnIκB or dnTCF4, and were either left untreated, or were treated with IL-1 as indicated. Cells were also transfected with the FOP-FLASH reporter plasmid, and the results are presented as the ratio between TOP-FLASH and FOP-FLASH activity (Fig. 1D)
dnTCF4 did not interfere with the ability of THP1 macrophages (Fig. 1A) or IL-1 (Fig. 1C) to induce NF-κB activity, demonstrating that Wnt signaling does not contribute to IL-1 mediated NF-κB activation. This experiment also demonstrated that in our system, Wnt/β-catenin signaling does not inhibit the ability of THP1 macrophages (Fig. 1A), IL-1 (Fig. 1C), or TNF (not shown) to induce NF-κB activity, as has been recently reported [39]. As expected, transfection of HCT116 cells with dnIκB prevented the ability of IL-1 to activate NF-κB (Fig. 1C) and IL-1 induced Wnt signaling was abolished in cells transfected with dnTCF4 (Fig. 1D).
To determine whether IL-1 activates Wnt signaling in a NF-κB dependent manner, we transfected HCT116 cells with the TOP-FLASH and FOP-FLASH reporter vectors in the presence of dnIκB. In cells transfected with an empty plasmid (neo), IL-1 induced ~ 3-fold increase in TOP/FOP activity (Fig. 2A). In contrast, expression of dnIκB abolished the ability of IL-1β to induce Wnt signaling, demonstrating that NF-κB activity is required for IL-1 to induce Wnt signaling (Fig. 2A).

IL-1 and macrophages induce Wnt signaling in a NF-κB dependent manner. a HCT116 cells were transiently transfected with the TOP-FLASH reporter gene together with an empty vector (neo), or dnIκB, and were treated with IL-1 as indicated. The results are presented as the ratio between the TOP-FLASH and FOP-FLASH activity. b HCT116 cells were transfected with the TOP-FLASH reporter gene together with an empty vector (neo), dnIκB, or dnTCF4 and were cultured with normal human monocytes (Mo) or THP1 macrophages for 24 h. C: Cell lysates from cells transfected with an empty vector (neo) or dnIκB were examined for the expression of pGSK3β and active β-catenin
We recently showed that colon cancer cells stimulate macrophages to release IL-1β (Kaler et al, in press). Consistent with this, normal human monocytes, precursors of the tumor associated macrophages, and THP1 macrophages were both potent inducers of Wnt signaling in tumor cells (Fig. 2B). On average, monocytes induced ~4-fold and ~THP1 macrophages ~ 3-fold increase in Wnt activity (Fig. 2B). However, like IL-1, macrophages failed to induce Wnt signaling in HCT116 cells transfected with dnIκB (Fig. 2B), and, as expected, in cells transfected with dnTCF4 (Fig. 2B). These data established that tumor associated macropohages induce Wnt signaling in tumor cells through a NF-κB dependent pathway.
To confirm the requirement of NF-κB activity for IL-1-induced Wnt/β-catenin signaling, we used antibody that specifically recognizes the phosphorylated, inactive form of GSK3β (pGSK3βSer9) to show that IL-1 and THP1 macrophages failed to inactivate GSK3β in HCT116 cells expressing dnIκB, and that the levels of active (unphosphorylated) β-catenin were significantly reduced in HCT116 cells with impaired NF-κB signaling (Fig. 2C).
IL-1 and Macrophages Activate PDK1/ AKT Signaling in Tumor Cells
Because AKT has been shown to be a downstream target of NF-κB [40], to phosphorylate GSK3β [30] and to be involved in Wnt signaling [41,42], we next tested whether macrophages/IL-1 inactivate GSK3β through activation of the AKT in colon cancer cells. Serum starved HCT116 cells were either left untreated or were treated with IL-1 for 30 min, 1 h or 3 h, and cell lysates were examined for phosphorylation of AKT, using antibodies specific for AKT phosphorylated on Ser473 and Thr308. As shown in Fig. 3A, IL-1 treatment resulted in a rapid phosphorylation of AKT at both residues. Likewise, PDK1, a kinase responsible for activation of AKT, was activated by IL-1, and c-raf, a known target of AKT, was phosphorylated by IL-1 (Fig. 3A). In contrast, the levels of total AKT and β-actin were not modulated by the treatment with IL-1.

IL-1 and macrophages activate PDK1/AKT. a Serum starved HCT116 cells were treated with IL-1 as indicated and cell lysates were examined for phosphorylation of AKT, PDK1, GSK3β, and c-Raf. b HCT116 cells were cultured with peripheral blood monocytes (Mo) or THP1 macrophages and were examined for activation of AKT and phosphorylation of GSK3β. c HCT116 cells were cultured with peripheral blood monocytes either directly, or were co-cultured using transwell inserts (0.4 μm size). d HCT116 and Hke-3 cells were co-cultured with THP1 macrophages transfected with nontargeting siRNA (THP1) or siRNA specific for IL-1 or STAT1. The expression of pPDK1, pAKT, AKT and βactin was determined by immunoblotting
We showed that, like IL-1β, normal peripheral blood moncoytes and THP1 macrophages phosphorylate AKT and inactivate GSK3β in tumor cells (Fig. 3B). Monocytes were equally potent in inducing PDK1/AKT signaling when they were separated from the tumor cells with a cell impermeable membrane (Fig. 3C), confirming that they induce PDK1/AKT signaling in tumor cells through a soluble factor. To determine whether macrophages induce AKT signaling in tumor cells through IL-1, we co-cultured HCT116 and HKe-3 cells with THP1 macrophages with silenced IL-1β or STAT1, which we established is required for the IL-1 release from macrophages (Kaler et al, in press). We showed that IL-1 or STAT1 deficient THP1 macrophages failed to phosphorylate AKT or activate PDK1 in tumor cells (Fig. 3D), confirming that IL-1 mediates AKT dependent inactivation of GSK3β in tumor cells.
Finally, we showed that IL-1, THP1 macrophages and peripheral blood monocytes failed to phosphorylate AKT and PDK1 in tumor cells expressing dnIκB (Fig. 4A, data not shown), demonstrating that they activate AKT signaling in a NF-κB dependent manner. The NF-κB and AKT pathways are known to interact and AKT has been shown to be either downstream or upstream of NF-κB [29, 40]. We showed that transfection of cells with dnAKT (unlike transfection with dnIκB) did not impair the ability of macrophages, IL-1 or TNF to trigger IκBα degradation in HCT116 cells (Fig. 4B) and did not affect NF-κB transcriptional activity (data not shown), confirming that AKT acts downstream of NF-κB. This is consistent with our finding that macrophages and IL-1 failed to activate AKT in cells expressing dnIκB (Fig. 4A). The mechanism whereby NF-κB activates AKT phosphorylation is currently being investigated in the laboratory.

AKT acts downstream of NF-κB: a HCT116 cells were transfected with an empty plasmid (neo) or dnIκB and were cultured with THP1 macrophages or were treated with IL-1 as indicated. b HCT116 cells were transfected with an empty plasmid (neo), dnIκB, dnAKT or CA AKT and were treated as indicated. The levels of pAKT, pPDK1 and IκBα were determined by immunoblotting
AKT is Required for Macrophage and IL-1 Induced Wnt Signaling in Tumor Cells
To determine whether AKT is required for IL-1 induced Wnt signaling, we transfected HCT116 cells with the TOP-FLASH reporter plasmid in the absence or the presence of dnAKT. The expression of dnAKT was confirmed by immunoblotting with an anti HA antibody (Fig. 5C). We showed that the ability of IL-1 to induce Wnt signaling was abolished in cells transfected with dnAKT, demonstrating that AKT is required for the IL-1 mediated Wnt signaling (Fig. 5A). Consistently, normal peripheral blood monocytes and THP1 macrophages failed to induce Wnt signaling in tumor cells that were transfected with dnAKT (Fig. 5B), confirming that AKT mediates the crosstalk between tumor cells and macrophages. Consistent with the inability of IL-1 or THP1 macrophages to promote Wnt signaling in HCT116 cells transfected with dnAKT, these cells did not respond to IL-1 or THP1 macrophages with phosphorylation of GSK3β or activation of β-catenin (Fig. 5C). Finally, we showed that the expression of a constitutively active AKT (CA AKT) was sufficient to drive Wnt signaling (Fig. 5D).

AKT is required for IL-1 or macrophage-induced Wnt signaling. a and b HCT116 cells were transfected with the TOP-FLASH reporter gene and were co-transfected with an empty vector (neo) or dnAKT as indicated. Cells were left untreated (CTRL) or were treated with IL-1 or were co-cultured with normal human peripheral blood monocytes (Mo) or THP1 macrophages. c Cell lysates from HCT116 cells transfected with an empty vector (neo) or dnAKT were tested for the expression of pGSK3β and active β-catenin. The expression of dnAKT was confirmed by immunoblotting for HA. d HCT116 cells were transfected with the TOP-FLASH reporter gene together with increasing concentrations of an empty vector (neo) or constitutively active AKT (CA AKT). The expression of CA AKT was confirmed by immunoblotting for HA (see the inset). E: HCT116 cells were transfected with an empty plasmid (neo), dnIκB, dnAKT or CA AKT and were cultured with THP1 macrophages or were treated with IL-1 or TNF for 1 h. The levels of c-myc, c-myc Thr58/Ser62, c-jun and βactin were determined by immunoblotting
We showed previously that macrophages and IL-1 induce the expression of Wnt target genes in tumor cells, including c-myc (Kaler et al, in press). c-Myc activity is also regulated at the posttranslational level through GSK3β mediated inhibitory phosphorylation of c-myc at Thr58, and ERK activating phosphorylation at Ser62 [43]. We demonstrated that macrophages and IL-1 induced c-myc phosphorylation on Thr58/Ser62 in tumor cells (Fig. 5E), demonstrating that factors in the tumor microenvironment also regulate the stability of Myc protein in tumor cells.
The ability of THP1 macrophages and IL-1 to induce the expression of c-myc and c-jun and to increase c-myc phosphorylation was abrogated not only in tumor cells transfected with dnIκB (Fig. 5E), but also in cells transfected with dnAKT (Fig. 5F), confirming the requirement of AKT for Wnt signaling. The expression of CA AKT was not sufficient to significantly increase the basal expression of c-myc or c-jun, but it augmented the responsiveness of tumor cells to IL-1 and macrophages (Fig. 5F). TNF acted as a poor inducer of c-myc and c-jun, consistent with its weaker ability to induce Wnt signaling in HCT116 cells (not shown).
Altogether, these data established that IL-1/macrophages promote Wnt signaling and activate Wnt targets through activation of the NF-κB-dependent AKT signaling.
Pharmacological Inhibition of AKT by LY294002 or Taxotere Abrogates Wnt Signaling in Tumor Cells
To confirm the requirement of AKT for Wnt signaling, we tested whether pharmacological inhibition of AKT interferes with the ability of macrophages/IL-1 to promote Wnt signaling. HCT116 and Hke-3 cells transfected with the TOP-FLASH reporter vector were cultured with THP1 macrophages and were treated with IL-1 in the absence or the presence of LY294002 (LY), a specific inhibitor of PI3K/AKT signaling. While treatment of tumor cells with LY294002 did not modulate constitutive β-catenin/TCF driven transcriptional activity, it abrogated the ability of macrophages and IL-1 to induce Wnt signaling in both HCT116 and Hke-3 cells (Fig. 6), confirming that macrophages/IL-1 promote Wnt signaling in an AKT dependent manner.

Pharmacological inhibition of AKT by LY294002 or taxotere in HCT116 (a) and Hke-3 (b) cells inhibits enhanced Wnt signaling in tumor cells in response to macrophages or IL-1. Cells were transfected with the TOP-FLASH reporter gene and were cultured with THP1 cells or were treated with IL-1 in the presence of LY or taxotere as indicated. LY = LY294002 (20 μM), Tax = taxotere (10 nM)
Taxotere is a semi-synthetic analogue of taxol, which has been approved for the treatment of breast, ovarian, and non-small cell lung cancer. It inhibits the activity of AKT by promoting proteasomal degradation of the heat shock protein 90 (Hsp90) which protects AKT from dephosphorylation by PPA2 [44, 45]. Like LY294002, taxotere did not affect the basal Wnt signaling in either HCT116 or Hke-3 cells, but it abrogated the ability of macrophages and IL-1 to induce Wnt signaling in tumor cells (Fig. 6). These data confirmed that AKT mediates macrophages/IL-1 induced Wnt signaling and, moreover, demonstrate a novel mode of biological activity for taxotere.
Tumor Promoting Activity of Macrophages/IL-1 Require both NF-κB and AKT Signaling in Tumor Cells
We showed that macrophages and IL-1, through their ability to induce Wnt signaling, promote the clonogenic growth of colon cancer cells (Kaler et al, in press). Because we established that macrophages and IL-1 induce Wnt signaling in an NF-κB dependent manner (Fig. 2), we tested whether inhibition of NF-κB activity in tumor cells hampers the ability of macrophages and IL-1 to promote their growth. HCT116 cells were transfected with an empty vector or with dnIκB and the ability of THP1 macrophages or IL-1 to increase their clonogenic potential was examined as described in Material and Methods. As shown in Fig. 7A and B, while macrophages and IL-1 strongly increased the clonogenic growth of HCT116 cells transfected with an empty vector (neo), they failed to promote the growth of HCT116 cells with impaired NF-κB signaling. Macrophages and IL-1 also significantly increased the volume of the colonies in HCT116 cells, which was dependent on NF-κB signaling in tumor cells as well (Fig. 7B). Similar results were obtained in Hke-3 cells (not shown).

NF-κB (a, b) and AKT (c, d) mediate the growth promoting activity of macrophages: HCT116 cells were transfected with an empty vector (neo), dnIκB, or dnAKT as indicated, and were either cultured with macrophages or were stimulated with IL-1. The number of colonies and their volume were determined as described in Material and Methods. A and C show representative colonies. B: *, p < 0.0001, #, p < 0.0001: **,p = 0.013, ##, p = 0.022, D: *, p =< 0.0001, #, p = 0.0001: **, p = 0.0003, ##, p = 0.00003
Since we demonstrated that AKT is downstream of NF-κB, we next tested whether inhibition of AKT activity in tumor cells alters their interactions with macrophages. Macrophages and IL-1 increased both the size and the number of colonies in HCT116 cells transfected with an empty vector, but not in cells transfected with dnAKT (Fig. 7C and D), demonstrating that AKT mediates the growth promoting activity of macrophages/IL-1. In two independent experiments, each performed in duplicate, both HCT116 and Hke-3 cells transfected with dnAKT yielded colonies with significantly larger volume; the reason for this increase remains, for now, unknown.
In summary, our data demonstrate that, as tumor cells recruit normal peripheral blood monocytes to the tumor microenvironment, they stimulate them to release IL-1β. We showed that tumor associated macrophages and recombinant IL-1 exert their protumorigenic activity through NF-κB/AKT dependent activation of Wnt signaling in tumor cells (Fig. 8), establishing a novel molecular link among inflammation, Wnt signaling and tumor progression.

Signaling pathway whereby tumor associated macrophages promote Wnt signaling in tumor cells. Peripheral blood monocytes (Mo) were cultured with control medium or with conditioned medium from HCT116 or Hke-3 cells for 48 h. As shown here, soluble factor(s) from HCT116 and Hke-3 cells induced maturation of normal peripheral blood monocytes (Mo), demonstrated by phalloidin/DAPI staining, coupled to the release of IL-1β. IL-1β, through activation of NF-κB, induced phosphorylation of PDK1 and AKT, which inactivates GSK3β, leading to enhanced β-catenin/TCF4 transcriptional activity, and increased expression of Wnt target genes in tumor cells, including c-myc and c-jun
Discussion
We recently reported that macrophages promote growth of colon cancer cells through IL-1 mediated, STAT1 dependent, activation of Wnt signaling (Kaler et al, in press). Here we show that peripheral blood monocytes, direct precursors of the tumor associated macrophages, and IL-1 activate Wnt signaling in tumor cells in a NF-κB/AKT dependent manner. NF-κB activity is triggered through activation of the IKK complex by a variety of proinflammatory cytokines, including TNF and IL-1, and activation of NF-κB in tumor cells appears to be a critical molecular link between inflammation and cancer [21, 46]. Specific inactivation of NF-κB signaling in intestinal cells dramatically decreased the incidence of intestinal tumors in a mouse model of colitis associated cancer [22]. Inhibition of NF-κB has been shown to convert LPS-induced growth of CT26 mouse colon carcinoma cells into LPS-induced tumor regression through apoptosis [23], demonstrating that an active NF-κB signaling in intestinal cells is required for tumor progression. Malignant cells have been shown to drive NF-κB activation in TAMs in order to maintain their immunosuppressive phenotype [47], suggesting that intact NF-κB signaling in both tumor cells and macrophages is required for the interaction of tumor cells with tumor associated macrophages. Here we show that macrophages and IL-1 failed to activate AKT signaling, inactivate GSK3β and to induce Wnt signaling in tumor cells with impaired NF-κB activity. Consistently, macrophages and IL-1 did not increase the clonogenic growth of colon cancer cells expressing dnIκB.
We established that NF-κB activity is required for macrophages and IL-1 to stimulate PDK1 and AKT in tumor cells, demonstrating that AKT is downstream of NF-κB signaling. The molecular link between the NF-κB and PDK1/AKT signaling remains to be determined, but both IL-1 and TNF have been shown to trigger AKT activation in a PI3K dependent manner [29, 48]. Several experiments indicate that AKT and Wnt signaling interact. It has been recently shown that nuclear AKT inhibits, whereas membrane tethered AKT stimulates β-catenin transcriptional activity [42], underscoring a complex nature of the crosstalk between the canonical Wnt and AKT signaling pathways. AKT has also been shown to directly phosphorylate β-catenin at Ser552, which, in contrast to the GSK-3β mediated pathway, does not alter β catenin stability, but promotes its nuclear translocation [41]. Thus, AKT can activate β-catenin/TCF transcriptional activity both by indirect stabilization of β-catenin through inhibition of GSK3β and by direct phoshorylation of β-catenin which promotes β-catenin nuclear accumulation. We demonstrated that IL-1 and tumor associated macrophages inactivate GSK3β in tumor cells, but do not have data to support a direct phosphorylation of β-catenin by IL-1 or by tumor associated macrophages.
The ability of macrophages and IL-1 to induce Wnt signaling and the expression of Wnt target genes, such as c-myc and c-jun, was abrogated in cells transfected with dnAKT. Consistently, macrophages and IL-1 failed to increase the clonogenic growth of tumor cells in the absence of AKT signaling, demonstrating that macrophages/IL-1 activate Wnt signaling and exert protumorigenic activity through a NF-κB/AKT dependent pathway.
Several of our experiments were performed with the transformed THP1 monocytic cell line, however we have confirmed that normal peripheral blood monocytes, cells that are recruited to the tumor microenvironment and polarize into tumor associated macrophages, also secrete IL-1 when stimulated with tumor cells, induce Wnt signaling and expression of Wnt target genes in tumor cells and promote tumor growth (Kaler et al, in press). Here we showed that, like THP1 cells, normal monocytes promote Wnt signaling in tumor cells through an NF-κB (Fig. 2B) and AKT (Fig. 5B) dependent pathway.
Abnormal activation of AKT is found in a variety of human tumors, including colorectal cancer, as a result of activating mutations of PIK3CA, overexpression of AKT, the loss of PTEN, or constitutive signaling by Ras [49]. However, it was demonstrated that in epithelial cells mutant Ras is not sufficient for full activation of the PI3K kinase, induction of AKT or inactivation of GSK3β [50] and that co-expression of Ras with PIK3CA is required for AKT activation and full transformation. Consistently, colorectal tumors often co-express kRas and PI3KCA mutations [51]. However, despite the fact that HCT116 cells carry both kRas mutation and the PI3KCA mutation [52], the level of activated AKT in these cells is rather low (Fig. 3). We showed that tumor associated macrophages, or IL-1, significantly increase AKT signaling in HCT116 cells and inactivate GSK3β, suggesting that inflammatory signals may substitute for the cooperative mutations during tumor progression.
A number of studies have established that inflammation contributes to many types of malignancies, including colorectal cancer. Consistently, IBD patients have elevated risk for colorectal cancer, and anti-inflammatory agents exert chemopreventive activity. Mutations in NOD2 that have been linked to Crohn’s disease, and therefore to increased risk of colorectal cancer, are associated with increased production of IL-1β and increased colonic inflammation [53]. The role of NF-κB, which is a major signaling pathway utilized by proinflammatory cytokines, including IL-1β, in ulcerative colitis and colon cancer has been established [22]. In this report we present data which demonstrate that IL-1β-induced NF-κB activation is coupled to Wnt signaling, a major oncogenic pathway which regulates differentiation and proliferation of intestinal epithelial cells. Our findings established a direct link between inflammation and tumor progression, and suggest a model whereby Wnt driven tumorigenesis is modulated by IL-1β-dependent signaling from the macrophages present in the tumor microenvironment.
Colon cancer development/progression can be controlled by chemopreventive agents, such as nonsteroidal anti-inflammatory drugs (NSAIDs) and vitamin D. NSAIDs act through inhibition of COX-2 activity [54] and inhibition of peroxisome proliferator-activated receptor δ (PPARδ) [55]. Several NSAIDs, such as sulindac and aspirin, are also potent inhibitors of NF-κB activity in tumor cells [56,57]. Because we showed that NF-κB signaling mediates the interaction of tumor cells with tumor associated macrophages, our results suggest that NSAIDs may exert their chemopreventive activity also by interrupting a crosstalk between the tumor cells and the tumor associated macrophages. We have recently reported that vitamin D, another potent chemopreventive agent for colon cancer, alters the ability of macrophages to promote tumor growth through inhibition of the release of IL-1 from macrophages (Kaler et al, in press). Likewise, our data suggest that inhibitors of PI3K/AKT signaling, which are in preclinical and clinical trials, may also interrupt the crosstalk between the tumor cells and stroma. Our demonstration that taxotere, an inhibitor of AKT activity, hampers the ability of macrophages to induce Wnt signaling in tumor cells provides support for such a premise.
Thus, it appears that commonly used chemopreventive and chemotherapeutic agents can prevent tumor progression by disrupting the interaction of tumor cells with the tumor microenvironment, acting either on the tumor cells themselves, or on the cells in the tumor microenvironmet.
References
Mantovani A, Sica A, Sozzani S et al (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23:549–555
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4:71–78
Brabletz T, Jung A, Hermann K et al (1998) Nuclear overexpression of the oncoprotein beta-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res Pract 194:701–704
Brabletz T, Jung A, Reu S et al (2001) Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA 98:10356–10361
Eberhard A, Kahlert S, Goede V et al (2000) Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res 60:1388–1393
Goede V, Brogelli L, Ziche M, Augustin HG (1999) Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int J Cancer 82:765–770
Goede V, Fleckenstein G, Dietrich M et al (1998) Prognostic value of angiogenesis in mammary tumors. Anticancer Res 18:2199–2202
Hanada T, Nakagawa M, Emoto A et al (2000) Prognostic value of tumor-associated macrophage count in human bladder cancer. Int J Urol 7:263–269
Lissbrant IF, Stattin P, Wikstrom P et al (2000) Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int J Oncol 17:445–451
Salvesen HB, Akslen LA (1999) Significance of tumour-associated macrophages, vascular endothelial growth factor and thrombospondin-1 expression for tumour angiogenesis and prognosis in endometrial carcinomas. Int J Cancer 84:538–543
Etoh T, Shibuta K, Barnard GF, Kitano S, Mori M (2000) Angiogenin expression in human colorectal cancer: the role of focal macrophage infiltration. Clin Cancer Res 6:3545–3551
Forssell J, Oberg A, Henriksson ML et al (2007) High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res 13:1472–1479
Oosterling SJ, van der Bij GJ, Meijer GA et al (2005) Macrophages direct tumour histology and clinical outcome in a colon cancer model. J Pathol 207:147–155
Krelin Y, Voronov E, Dotan S et al (2007) Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res 67:1062–1071
Apte RN, Voronov E (2008) Is interleukin-1 a good or bad ‘guy’ in tumor immunobiology and immunotherapy? Immunol Rev 222:222–241
Voronov E, Shouval DS, Krelin Y et al (2003) IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci USA 100:2645–2650
Vidal-Vanaclocha F, Fantuzzi G, Mendoza L et al (2000) IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci USA 97:734–739
Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z (1997) MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity 7:837–847
Rakoff-Nahoum S, Medzhitov R (2007) Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 317:124–127
Karin M (2006) Nuclear factor-kappaB in cancer development and progression. Nature 441:431–436
Greten FR, Eckmann L, Greten TF et al (2004) IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118:285–296
Luo JL, Maeda S, Hsu LC, Yagita H, Karin M (2004) Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell 6:297–305
Alessi DR, James SR, Downes CP et al (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7:261–269
Vanhaesebroeck B, Alessi DR (2000) The PI3K-PDK1 connection: more than just a road to PKB. Biochem J 346(Pt 3):561–576
Datta SR, Dudek H, Tao X et al (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241
Cardone MH, Roy N, Stennicke HR et al (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282:1318–1321
Brunet A, Bonni A, Zigmond MJ et al (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868
Ozes ON, Mayo LD, Gustin JA et al (1999) NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 401:82–85
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789
Ikeda S, Kishida S, Yamamoto H et al (1998) Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. Embo J 17:1371–1384
Kishida S, Yamamoto H, Ikeda S et al (1998) Axin, a negative regulator of the wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of beta-catenin. J Biol Chem 273:10823–10826
Moon RT, Bowerman B, Boutros M, Perrimon N (2002) The promise and perils of Wnt signaling through beta-catenin. Science 296:1644–1646
Van der Flier LG, Sabates-Bellver J, Oving I et al (2007) The Intestinal Wnt/TCF Signature. Gastroenterology 132:628–632
Shirasawa S, Furuse M, Yokoyama N, Sasazuki T (1993) Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 260:85–88
DiDonato J, Mercurio F, Rosette C et al (1996) Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol 16:1295–1304
Franke TF, Yang SI, Chan TO et al (1995) The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81:727–736
Klampfer L, Huang J, Shirasawa S, Sasazuki T, Augenlicht L (2007) Histone Deacetylase Inhibitors Induce Cell Death Selectively in Cells That Harbor Activated kRasV12: The Role of Signal Transducers and Activators of Transcription 1 and p21. Cancer Res 67:8477–8485
Deng J, Miller SA, Wang HY et al (2002) beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell 2:323–334
Meng F, Liu L, Chin PC, D’Mello SR (2002) Akt is a downstream target of NF-kappa B. J Biol Chem 277:29674–29680
Fang D, Hawke D, Zheng Y et al (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282:11221–11229
Li FQ, Mofunanya A, Harris K, Takemaru K (2008) Chibby cooperates with 14–3-3 to regulate beta-catenin subcellular distribution and signaling activity. J Cell Biol 181:1141–1154
Sears R, Nuckolls F, Haura E et al (2000) Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 14:2501–2514
Murtagh J, Lu H, Schwartz EL (2006) Taxotere-induced inhibition of human endothelial cell migration is a result of heat shock protein 90 degradation. Cancer Res 66:8192–8199
Sato S, Fujita N, Tsuruo T (2000) Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci USA 97:10832–10837
Lin WW, Karin M (2007) A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 117:1175–1183
Hagemann T, Lawrence T, McNeish I et al (2008) “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med 205:1261–1268
Cahill CM, Rogers JT (2008) Interleukin (IL) 1beta induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/IkappaB kinase alpha pathway targeting activator protein-1. J Biol Chem 283:25900–25912
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2:489–501
Oda K, Okada J, Timmerman L et al (2008) PIK3CA cooperates with other phosphatidylinositol 3′-kinase pathway mutations to effect oncogenic transformation. Cancer Res 68:8127–8136
Parsons DW, Wang TL, Samuels Y et al (2005) Colorectal cancer: mutations in a signalling pathway. Nature 436:792
Jhawer M, Goel S, Wilson AJ et al (2008) PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res 68:1953–1961
Maeda S, Hsu LC, Liu H et al (2005) Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science 307:734–738
Gupta RA, Dubois RN (2001) Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer 1:11–21
He TC, Chan TA, Vogelstein B, Kinzler KW (1999) PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 99:335–345
O’Neill EA (1998) A new target for aspirin. Nature 396:15 17
Yin MJ, Yamamoto Y, Gaynor RB (1998) The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396:77–80
Acknowledgments
We thank Dr. Anna Velcich for reading the manuscript. Supported in part by CA 111361, U54 CA 100926 and P30-13330 from NCI.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kaler, P., Godasi, B.N., Augenlicht, L. et al. The NF-κB/AKT-dependent Induction of Wnt Signaling in Colon Cancer Cells by Macrophages and IL-1β. Cancer Microenvironment 2, 69–80 (2009). https://doi.org/10.1007/s12307-009-0030-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-009-0030-y