
Abstract
Recent studies aimed at elucidating the mechanism controlling HIV-1 transcription have led to the identification and characterization of two multi-subunit complexes that both contain P-TEFb, a human transcription elongation factor and co-factor for activation of HIV-1 gene expression by the viral Tat protein. The first complex, termed the 7SK snRNP, acts as a reservoir where active P-TEFb can be withdrawn by Tat to stimulate HIV-1 transcription. The second complex, termed the super elongation complex (SEC), represents the form of P-TEFb delivered by Tat to the paused RNA polymerase II at the viral long terminal repeat during Tat transactivation. Besides P-TEFb, SEC also contains other elongation factors/co-activators, and they cooperatively stimulate HIV-1 transcription. Recent data also indicate SEC as a target for the mixed lineage leukemia (MLL) protein to promote the expression of MLL target genes and leukemogenesis. Given their roles in HIV-1/AIDS and cancer, further characterization of 7SK snRNP and SEC will help develop strategies to suppress aberrant transcriptional elongation caused by uncontrolled P-TEFb activation. As both complexes are also important for normal cellular gene expression, studying their structures and functions will elucidate the mechanisms that control metazoan transcriptional elongation in general.
Similar content being viewed by others

Transcriptional elongation as a major gene expression control point
Transcription by RNA polymerase II (RNAPII) can be subdivided into multiple stages, of which the initiation and elongation stages are considered the two primary targets for controlling eukaryotic gene expression (Kuras and Struhl 1999; Saunders et al. 2006; Core and Lis 2008). However, during most of the past three decades, attention of the transcription field had been largely focused on the initiation stage, as the recruitment of RNAPII to a few model gene promoters was found to be the major rate-limiting step for their transcription (Kuras and Struhl 1999; Ptashne 2005). In comparison, transcriptional control at the elongation stage had been generally neglected and viewed as a mechanism that is used by only a few selected genes and under highly specialized conditions.
A major paradigm shift in the transcription field occurred in 2007 when global analyses conducted in both Drosophila and human stem cells revealed that a large number of genes that often play important roles in controlling cell growth, renewal, and differentiation have paused RNAPII at their promoter-proximal regions even under resting, un-stimulated conditions (Guenther et al. 2007; Muse et al. 2007). For these genes, transcriptional activation does not involve the recruitment of RNAPII and setting up a pre-initiation complex at the promoters, which can be very time-consuming. Rather, the transition of RNAPII from its promoter-proximal paused state into highly productive elongation mode is the defining moment of their activation. Because these genes are already in a state of suspended transcription prior to activation, the subsequent induction of RNAPII elongation can proceed very rapidly, thus allowing highly sensitive and synchronous response that is essential for cell growth and developmental control. The widespread existence of paused RNAPII in metazoan genomes suggests that transcriptional elongation plays a much more prominent and general role in regulating gene expression than previously appreciated.
Tat activation of HIV-1 transcriptional elongation
The detection of promoter-proximal pausing of RNAPII at many gene promoters was indeed a major breakthrough in the transcription field at large. However, to those who had been studying HIV-1 gene expression control, the discovery came as no great surprise. Prior to the recognition of the general importance of elongation control, HIV-1 transcription had long been known to be regulated exquisitely at the elongation stage (Kao et al. 1987). In fact, HIV-1 had been used as a favored model system to study this phenomenon. As such, our understanding of elongation control has benefited greatly from studies of HIV-1 and its activation by a combination of viral and host transcription factors.
Unlike simpler retroviruses that rely almost exclusively on the host cellular machinery for replication, lentiviruses, of which HIV-1 is a prime example, encode additional accessory proteins that further control the viral life cycle. The transcriptional transactivator (Tat) is one such key accessory protein encoded by HIV-1. During active infection, Tat is expressed early after the proviral DNA integrates into the host genome. Without Tat, RNAPII has been found to clear the HIV-1 long terminal repeat (LTR) successfully but soon pause, producing only short viral transcripts (Kao et al. 1987). Ample evidence indicates that Tat does not act alone and must cooperate with host cellular co-factors to activate HIV-1 transcription. After many years of effort aimed at elucidating the mechanism of Tat transactivation, the late 1990s finally saw the identification of the human positive transcription elongation factor b (P-TEFb) as a specific and essential human co-factor for Tat function (Mancebo et al. 1997; Zhu et al. 1997; Wei et al. 1998).
Consisting of the cyclin-dependent kinase 9 (CDK9) and its regulatory partner cyclin T1 (CycT1; other minor cyclin forms also exist but do not interact with Tat), P-TEFb is recruited to the HIV-1 LTR through interacting with Tat and the transactivation response (TAR) element, an RNA stem-loop structure formed by the nascent 5′ end of viral transcripts that are synthesized before RNAPII pauses (for reviews, see Peterlin and Price 2006; Zhou and Yik 2006). Once positioned next to the paused polymerase, the CDK9 kinase phosphorylates its primary substrates, the C-terminal domain (CTD) of the largest subunit of RNAPII and a pair of negative transcription elongation factors, DSIF and NELF. These phosphorylation events antagonize the actions of the negative elongation factors and release RNAPII from promoter-proximal pausing, leading to the production of full-length viral transcripts (Peterlin and Price 2006; Zhou and Yik 2006).
Cellular control of P-TEFb activity
P-TEFb is not a transcription elongation factor made just for HIV-1. Rather, its activity is also important for the expression of a vast majority of cellular genes in uninfected cells (Chao and Price 2001). Recent whole genome analyses have shown that inhibiting P-TEFb activity prevents the release of paused RNAPII at most active gene loci in embryonic stem cells (Rahl et al. 2010), again underlining P-TEFb’s general role in transcription.
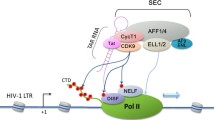
Given the importance of transcriptional elongation in controlling both HIV-1 and cellular gene expression and a key role for P-TEFb in this process, one can expect that the activity of P-TEFb is tightly regulated in the cell in order to optimally address the transcriptional needs of both the virus and its host. Indeed, P-TEFb has been shown to interact with a variety of protein and RNA regulators, and these interactions dynamically modulate the level of active P-TEFb available for HIV-1 and cellular gene expression (Fig. 1; Zhou and Yik 2006). For example, under normal growth conditions, more than half of the P-TEFb population in the HeLa nucleus are sequestered in a catalytically inactive complex termed the 7SK snRNP that also contains the 7SK snRNA and nuclear proteins HEXIM1 (or the homologous HEXIM2), LARP7, and MePCE (Fig. 1; Nguyen et al. 2001; Yang et al. 2001; Yik et al. 2003; Jeronimo et al. 2007; He et al. 2008). Within this complex, 7SK, an evolutionally conserved snRNA transcribed by RNA polymerase III, is protected against exonuclease cleavage by the actions of MePCE, the 7SK methylphosphate capping enzyme, as well as LARP7, a La-related protein associated with the 3’-poly(U) track of 7SK (Jeronimo et al. 2007; He et al. 2008; Xue et al. 2010). In return, 7SK functions as a molecular scaffold to maintain the integrity of 7SK snRNP, which sequesters P-TEFb and allows the CDK9 kinase activity to be inhibited by HEXIM1/2 in a 7SK-dependent manner (Fig. 1; Yik et al. 2003; Michels et al. 2004).

The 7SK snRNP is a reservoir of nuclear P-TEFb that can be withdrawn for activated transcription in response to HIV-1 infection, stress and other growth-stimulating signals. In the nucleus, a major portion of P-TEFb is sequestered in the 7SK snRNP, where the integrity of 7SK snRNA is maintained by MePCE and LARP7 and P-TEFb’s kinase activity is inhibited by HEXIM1 in a 7SK-dependent manner. Under a number of conditions including HIV-1 infection, certain stress treatments, exposure of cardiac myocytes to hypertrophic signals and activation of TCR in T cells, 7SK snRNP is disrupted to release P-TEFb for transactivation of P-TEFb-dependent genes. When P-TEFb is released during HIV-1 infection, it joins the viral Tat protein and several other host cellular proteins whose identification and characterization will be discussed below. Under other conditions that disrupt the 7SK snRNP, P-TEFb is picked up by Brd4 and delivered to a chromatin template to stimulate general transcriptional elongation. Contrary to those stress and growth-promoting signals, treating murine erythroleukemia cells with the differentiation-inducer HMBA shifts the P-TEFb equilibrium to the HEXIM1/7SK-bound state. A low level of 7SK snRNP has also been detected on the HIV-1 LTR under basal, un-stimulated conditions although its physiological significance is yet-to-be determined
The nuclear level of 7SK snRNP undergoes dynamic changes under a variety of conditions that globally affect cell growth and differentiation (Fig. 1). For example, the exposure of cardiac myocytes to hypertrophic signals triggers the release of P-TEFb from 7SK snRNP, leading to an overall increase in cellular protein and RNA contents and hypertrophic growth (Sano et al. 2002; Huang et al. 2004). Moreover, co-stimulating Jurkat T cells with anti-CD3/anti-CD28 antibodies to activate the T cell receptor (TCR) pathway also results in the disruption of 7SK snRNP and liberation of P-TEFb (Natarajan et al. 2010). When cells are treated with stress-inducing agents such as DNA-damaging agents actinomycin D and UV irradiation, and the kinase inhibitors DRB (5,6-dichloro-1-β-d-ribofuranosylbenzimidazole), flavopiridol, staurosporine, and H7 (1-(5-isoquinolinesulfonyl)-2-methylpiperazine), P-TEFb has also been found to dissociate from 7SK snRNP to mediate the stress-induced HIV-1 and cellular gene expression (Nguyen et al. 2001; Yang et al. 2001; Chen et al. 2004; Biglione et al. 2007). Finally, the 7SK snRNP level in murine erythroleukemia cells (MELC) shows a biphasic response upon the exposure to hexamethylene bisacetamide (HMBA; He et al. 2006), an inducer of MELC differentiation. During the initial 1–2 h, HMBA induces a transient disruption of 7SK snRNP, which is then followed by a permanent increase in the levels of HEXIM1 and 7SK snRNP after a prolonged treatment (He et al. 2006). Taken together, all these observations are consistent with the notion that the 7SK snRNP serves as a reservoir, from which active P-TEFb can be withdrawn in response to increased demands for elevated gene expression during active cell growth and response to environmental changes/stress (Zhou and Yik 2006).
Brd4 recruitment of P-TEFb for general transcription but not Tat transactivation
Once P-TEFb is released from the 7SK snRNP, it often finds its way into another complex that is characterized by the presence of the bromodomain protein Brd4 (Fig. 1; Jang et al. 2005; Yang et al. 2005). Brd4 is a ubiquitously expressed nuclear protein belonging to the bromodomain and ET domain family of proteins that contain two N-terminal tandem bromodomains and an extraterminal domain (Jeanmougin et al. 1997; Dey et al. 2000). While the motif located near the C terminus of Brd4 has been shown to be responsible for binding to P-TEFb (Bisgrove et al. 2007), the two bromodomains residing in the N-terminal region of Brd4 are involved in the interaction with the acetylated tails of histone H3 and H4 (Dey et al. 2003). Notably, the association of Brd4 with acetylated chromatin can persist through mitosis (Dey et al. 2000; Dey et al. 2003; Yang et al. 2008). These properties enable Brd4 to recruit P-TEFb to a chromatin template, beginning around mid- to late anaphase and before nuclear envelope/lamina formation and nuclear import of other general transcription factors (Yang et al. 2008). This leads to activation of the expression of P-TEFb-dependent genes in the early G1 phase of the cell cycle (Mochizuki et al. 2008; Yang et al. 2008). As such, Brd4 has been proposed to play a key role in transmitting epigenetic memory across cell division (Dey et al. 2003; Loyola and Almouzni 2004).
The recruitment of P-TEFb by Brd4 is likely to be important for general transcriptional elongation of both cellular and viral genes including basal, Tat-independent HIV-1 transcription (Fig. 1; Jang et al. 2005; Yang et al. 2005; Hargreaves et al. 2009). However, a number of sequence-specific transcriptional activators (e.g., HIV-1 Tat, NF-κB, Myc, CIITA, etc.) have been shown to interact with P-TEFb (Barboric et al. 2001; Eberhardy and Farnham 2001; Kanazawa et al. 2003; Kanazawa et al. 2000), and they could potentially bypass the requirement for Brd4 and deliver P-TEFb directly to their respective target genes (Zhou and Yik 2006). An excellent example to illustrate this point is provided by HIV-1 Tat. Evidence has been presented to show that the recruitments of P-TEFb by Tat and Brd4 are two mutually exclusive events that cannot occur at the same time (Yang et al. 2005; Bisgrove et al. 2007). In fact, Brd4 interferes with Tat transactivation as it competes with Tat for binding to P-TEFb (Yang et al. 2005). Furthermore, it has been shown that overexpression of the P-TEFb-interacting domain located at the C terminus of Brd4 disrupts the Tat-P-TEFb interaction and inhibits Tat transactivation and TNF-α-induced reactivation of latent HIV-1 (Bisgrove et al. 2007).
Where does Tat get its P-TEFb?
The demonstrations that a major source of nuclear P-TEFb exists in the 7SK snRNP raise the questions of where exactly Tat obtains its P-TEFb. The answer to this question has come from several independent studies all showing that Tat triggers the release of P-TEFb from 7SK snRNP in vitro and in vivo (Fig. 1; Schulte et al. 2005; Barboric et al. 2007; Sedore et al. 2007). Consistently, primary blood lymphocytes display a reduced amount of nuclear 7SK snRNP upon HIV-1 infection (Barboric et al. 2007). The existence of multiple structural and sequence similarities between the two P-TEFb-containing ribonucleoprotein (RNP) complexes, with one containing the HIV-1 TAR RNA and the other the cellular 7SK snRNA, likely contributed to this phenomenon (Zhou and Yik 2006).
However, the exact mechanism used by Tat to extract P-TEFb from 7SK snRNP remains controversial. Several published studies show that this ability of Tat depends on the integrity of its N-terminal activation domain and stems from the high affinity interaction between Tat and CycT1, which allows Tat to directly displace HEXIM1 from CycT1 (Schulte et al. 2005; Barboric et al. 2007; Sedore et al. 2007; Krueger et al. 2010). Once P-TEFb leaves the complex, a conformational change in 7SK blocks re-association of HEXIM1 (Krueger et al. 2010).
Revealing a different mechanism used by Tat to capture P-TEFb from the 7SK snRNP, a recent study implicates the Arginine-rich TAR-binding domain of Tat as critical in this process (Muniz et al. 2010). Tat is shown to use this domain to interact with a portion of the 7SK snRNA, which is normally contacted by HEXIM1 but structurally similar to the Tat-binding site of HIV-1 TAR, and cause the release of P-TEFb (Muniz et al. 2010). What remains to be seen from this study is how the observed Tat-7SK interaction will eventually be turned into the Tat/TAR/P-TEFb-containing complex that is necessary for Tat transactivation.
Another possible mechanism by which Tat extracts P-TEFb from 7SK snRNP involves the use of protein phosphatase 1 (PP1). This enzyme has been demonstrated to play a key role in stress-induced disruption of 7SK snRNP through dephosphorylating Threonine186 located at the tip of the CDK9 T-loop (Chen et al. 2008). Interestingly, Tat has been shown to bind PP1 directly (Ammosova et al. 2005), which can presumably deliver the enzyme to the 7SK snRNP to induce the latter’s disruption. Consistent with this notion, inhibition of PP1 in cultured cells is reported to block Tat activation of HIV-1 transcription and replication, and at the same time, increase the nuclear 7SK snRNP level (Ammosova et al. 2011). Since the engagement of PP1 by Tat will likely lead to the release of P-TEFb with the dephosphorylated CDK9 T-loop and thus catalytically inactive, it is postulated that there must be a subsequent, yet-to-be defined re-phosphorylation step to return P-TEFb to its active state prior to its contribution to Tat activation of HIV-1 transcription (Chen et al. 2008).
In addition to investigating the mechanisms of Tat disruption of 7SK snRNP, recent efforts have also been focused on determining the subnuclear location where this event takes place. In binding studies conducted in vitro, HEXIM1 has been shown to bind to the HIV-1 TAR RNA and inhibit P-TEFb kinase activity (Sedore et al. 2007), implicating a possible association of the 7SK snRNP with the LTR through TAR. Using the chromatin immunoprecipitation assay, another study also detects the association of 7SK snRNP with the pre-initiation complex formed on the HIV-1 LTR (D'Orso and Frankel 2010). However, this association is shown to proceed in the absence of TAR RNA, and the synthesis of TAR actually triggers the release of P-TEFb for activated HIV-1 transcription (D'Orso and Frankel 2010). Although it is quite clear that 7SK snRNP can indeed be found on the LTR during basal transcription (as indicated on the ∆TAR template), its level appears to be very low and cannot fully account for the high P-TEFb level detected on the LTR upon Tat activation (D'Orso and Frankel 2010), suggesting that the bulk of P-TEFb required for activated transcription may come from a different route.
Obviously, more studies are needed to fully understand the physiological significance of the association of 7SK snRNP with the HIV-1 LTR (Fig. 1) and determine exactly how and where the complex is targeted by Tat. Nevertheless, it is abundantly clear from the published data that Tat has the ability to not only recruit P-TEFb to the LTR but also increase the pool of functional P-TEFb for HIV-1 transcription through actively extracting P-TEFb from the 7SK snRNP.
Tat promotes the formation of a novel P-TEFb-containing complex and recruits it to the HIV-1 LTR
Once P-TEFb is released from the 7SK snRNP, it had not been clear whether it joins Tat on the HIV-1 LTR all by itself or together with other yet-to-be identified factors (Fig. 1). To address this question, a tandem affinity-purification approach employing anti-Flag and then anti-HA beads was used to purify the complex that contains both HA-tagged Tat and Flag-tagged CDK9 (Fig. 2; He et al. 2010). Although numerous cellular factors that can bind to either Tat or P-TEFb separately had been identified in the past, this was the first attempt to specifically isolate factors that are integral components of a complex(es) that contains both proteins (Fig. 2). Analyses of the purified materials by mass spectrometry reveal that in addition to Tat, CDK9 and CycT1, the Tat-P-TEFb complex also contains ELL2, AFF4, ENL, and AF9 (He et al. 2010). Importantly, the binding of these factors to Tat and P-TEFb has also been independently confirmed by Sobhian et al. (2010). Through directly isolating the Tat-associated proteins, the latter study has also identified several additional proteins (Sobhian et al. 2010). However, CDK9, CycT1, ELL2, AFF4, ENL and AF9 are likely the core subunits of a single multi-component complex now called the super elongation complex (SEC; He et al. 2010; Lin et al. 2010; Sobhian et al. 2010).

Tandem affinity-purification of SEC. Left, the HEX293-based TTAC cell line that stably expresses CDK9-Flag (CDK9-F) and conditionally expressing Tat-HA upon the induction by doxycycline from transduced retroviral vectors has been established. CDK9-F and Tat-HA interact with their natural partners to form various complexes in the nucleus, and the one containing both proteins is the target of purification. Right, in the first step of purification, nuclear extract prepared from TTAC cells is incubated with the anti-Flag monoclonal antibody (mAb) beads, which specifically attract CDK9-F and its associated factors. After extensive washes, CDK9-F and its associated factors are eluted off the beads with a buffer containing synthetic Flag peptide. In the second step, the materials derived from the first step are subjected to further purification with the anti-HA beads to yield the purified SEC complex containing both CDK9-F and Tat-HA and other associated factors
Like P-TEFb, the SEC subunit ELL2 is a well-characterized elongation factor and a member of the ELL family of transcription factors (Shilatifard et al. 1997). Employing a mechanism different from that of P-TEFb, ELL2 stimulates elongation by increasing the catalytic rate and suppressing transient pausing of RNAPII (Shilatifard et al. 1996). AFF4, another SEC subunit, is a member of the AF4 family of transcription factors/co-activators. It is also recognized as a fusion partner of the mixed lineage leukemia (MLL) protein that causes infant acute lymphoblastic leukemia (Taki et al. 1999). Just like AFF4, the other two core SEC subunits ENL and AF9 are also fusion partners of MLL and involved in MLL-associated leukemia (Harper and Aplan 2008). Although AFF4, ENL, and AF9 have previously been reported to interact with P-TEFb and each other (Estable et al. 2002; Mueller et al. 2007; Mueller et al. 2009), this is the first time that they are specifically linked to HIV-1 Tat.
The association of Tat with SEC has been shown to serve two complementary purposes (Fig. 3). Firstly, Tat can recruit SEC that contains at least two well-established elongation factors of different classes to the viral LTR. This allows P-TEFb and ELL2 to act simultaneously on the same polymerase enzyme in a cooperative manner, which is most likely responsible for the powerful elongation activity attributed to Tat. Secondly, Tat has been shown to markedly promote SEC formation. ELL2 turns out to be a short-lived protein that is rapidly degraded by the proteasome (He et al. 2010). However, its stability can be greatly enhanced by the presence of Tat in a process that likely requires the CDK9 kinase activity (He et al. 2010). Tat-mediated ELL2 stabilization and accumulation in the cell allows more ELL2 to be sequestered into SEC, thus promoting SEC formation and SEC-dependent HIV-1 transcription.

Tat recruits SEC that contains two distinct classes of elongation factors to the HIV-1 LTR to synergistically activate viral transcription by RNAPII. Soon after transcription begins at the HIV-1 promoter, the progression of RNAPII is blocked by the concerted actions of negative elongation factors DSIF and NELF. This results in the production of a short RNA transcript that folds into a stem-loop structure called TAR. For RNAPII to escape from promoter-proximal pausing, the HIV-1 encoded Tat protein binds to the host cellular SEC complex that contains P-TEFb and ELL2, two well-known elongation factors of different classes, and transcriptional co-activators AFF4 and ENL or AF9. Tat then delivers SEC to the paused RNAPII through forming a stable complex on TAR RNA. Subsequently, while P-TEFb phosphorylates the RNAPII CTD as well as the negative elongation factors DSIF and NELF, ELL2 directly stimulates the catalytic rate of the same polymerase. This results in the synergistic activation of HIV-1 elongation and the production of full-length viral transcripts
Tat-independent formation and function of SEC
Just like P-TEFb, SEC does not exist exclusively for Tat transactivation. Besides being recruited by Tat for activated HIV-1 transcription, SEC is also targeted by MLL fusion proteins to induce aggressive acute leukemias in both children and adults (Mueller et al. 2009; Lin et al. 2010; Yokoyama et al. 2010). It is known that chromosomal translocations of the MLL gene and its partners can give rise to fusion proteins that cause misregulated expression of MLL target genes (e.g., the HOX genes) and uncontrolled proliferation of hematopoietic progenitors (Ayton and Cleary 2003; Cozzio et al. 2003). At least three subunits of SEC, namely AFF4, ENL, and AF9, are known as fusion partners of MLL (Slany 2009). When fused to the DNA-binding domain of MLL, these proteins have been demonstrated to deliver SEC and its powerful elongation stimulatory activity to the MLL target genes to promote leukemic transformation (Mueller et al. 2009; Lin et al. 2010; Yokoyama et al. 2010).
Not only is SEC essential for disease-associated transcriptional elongation mediated by Tat and MLL fusions, it also appears to be essential for normal transcriptional elongation of cellular genes (He et al. 2010; Lin et al. 2010; Sobhian et al. 2010). In cells that are free of HIV-1 infection and MLL-translocations, SEC can be detected readily (He et al. 2010). Under such conditions, the formation of SEC relies on AFF4, which functions as a scaffold to mediate the interactions of P-TEFb with the rest of SEC (He et al. 2010; Yokoyama et al. 2010). Moreover, AFF4 behaves like Tat to increase the half-life of ELL2, which in turn promotes ELL2’s incorporation into and formation of SEC (He et al. 2010).
Our recent unpublished data indicate that ENL and AF9, the two highly homologous core subunits of SEC, in fact exist in separate SECs that display similar but non-identical functions. In the absence of sequence-specific recruitment factors such as Tat and MLL, the evolutionarily conserved YEATS domain of ENL/AF9 targets SEC to chromatin by contacting the polymerase-associated factor complex (PAFc), and through PAFc, the paused Pol II. This explains why this domain is dispensable for leukemogenesis when ENL/AF9 is translocated to MLL (Slany et al. 1998; Yokoyama et al. 2010) whose DNA-binding activity likely substitutes for the chromatin-targeting function of the YEATS domain. Finally, contrary to popular belief, the histone lysine79 methyltransferase Dot1L, which is a well-known binding partner of ENL and AF9, competes with AFF4 for binding to ENL/AF9 and thus does not reside in SEC and also is unnecessary for SEC function.
SEC, 7SK snRNP, and the activation of HIV-1 latency
Latent reservoirs of HIV are the principal impediment to eradication of infection because they harbor integrated, transcriptionally silent proviruses that resume active infection once therapy is disrupted. Methods are currently being developed to rid of latent viral infection through reactivating these reservoirs, which can then be cleared by highly active antiretroviral therapy (HAART) and specific anti-HIV immunotoxins or viral cytopathogenicity (Margolis 2010; Trono et al. 2010). However, the most commonly employed chemical activators of latency are highly toxic and in need of major improvements to their specificity and efficacy (Marsden and Zack 2009). Since the repression of HIV-1 transcription plays a pivotal role in establishing viral latency in CD4+ T cells (Lassen et al. 2004; Contreras et al. 2006), the best-characterized HIV-1 reservoir, a better understanding of the mechanism and factors controlling HIV-1 transcription is crucial for the development of specific and efficient ways to bypass the requirement for the toxic activators and reactivate HIV-1.
Since P-TEFb is not recruited to the HIV-1 LTR in latently infected cells, this situation could potentially be exploited to reactivate the latent virus. Consistent with this idea, it has recently been shown that a restriction in P-TEFb activity due to its sequestration in the inactive 7SK snRNP in resting T cells contributes to the establishment of HIV-1 latency and can be overcome by activation of the TCR, which releases P-TEFb (Tyagi et al. 2010). Furthermore, HMBA and suberoylanilide hydroxamic acid, two compounds capable of activating latent HIV-1, have also been shown to release P-TEFb from 7SK snRNP (He et al. 2006; Contreras et al. 2007; Contreras et al. 2009), leading to the activation of viral replication in latently infected cell lines as well as primary T cells from HAART-treated patients (Vlach and Pitha 1993; Klichko et al. 2006; Contreras et al. 2007; Contreras et al. 2009).
The recent demonstration that the multi-subunit SEC complex represents the form of P-TEFb that is involved in both Tat-independent and Tat-dependent HIV-1 transcription (He et al. 2010; Sobhian et al. 2010) suggests the possibility of increasing the level and/or activity of SEC to activate latent viruses. Toward this goal, our data indicate that when the core SEC subunits ELL2 and AFF4 are co-expressed in a cell, ELL2 becomes stabilized and accumulates to a high level. This results in the synergistic and, more importantly, preferential activation of HIV-1 transcription to a level similar to that caused by HIV’s own Tat protein (He et al. 2010). Since the activation of latent HIV-1 must initially proceed in the absence of Tat, the ability of co-expressed ELL2 and AFF4 to mimic the Tat action underscores the importance to test the hypothesis that co-expressing these two proteins could provide an opportunity to activate viral replication in latently infected T cells.
Summary and perspectives
Human P-TEFb was first identified in 1997 as a specific host cellular co-factor for Tat activation of HIV-1 transcription (Mancebo et al. 1997; Zhu et al. 1997; Wei et al. 1998). Since then, this landmark discovery has provided the basic framework for our understanding of Tat function in the HIV-1 life cycle. Recently, the conventional view of HIV-1 gene expression control has been significantly expanded by the demonstrations that Tat captures P-TEFb from the 7SK snRNP, the major P-TEFb reservoir in the nucleus, promotes the formation of a novel P-TEFb complex termed SEC that also contains elongation factor ELL2 and several other transcription factors/co-factors, and delivers SEC to the paused RNAPII on the viral LTR (Fig. 3; He et al. 2010; Sobhian et al. 2010). The ability to allow P-TEFb and ELL2, representatives of two different classes of elongation factors, to act on the same polymerase enzyme explains why Tat is such a powerful transcriptional activator. These findings have not only provided fresh mechanistic insights into the control of HIV-1 gene expression but also revealed new targets for the development of improved anti-viral treatments as well as more specific and efficient therapeutic strategies to eradicate the latent HIV-1 reservoirs.
Despite these progresses, there are still a number of outstanding questions that remain to be answered. Firstly, the structures and functions of both 7SK snRNP and SEC await further characterization, which will enable us to better understand how and where Tat captures P-TEFb from 7SK snRNP and also the precise mechanism by which SEC stimulates HIV-1 and cellular transcriptional elongation. Furthermore, the relationship among several known P-TEFb-containing complexes requires further clarification. For example, it is known that the recruitment of P-TEFb by Brd4 occurs predominantly at the promoter region and is important for general transcriptional elongation (Jang et al. 2005; Yang et al. 2005). Once recruited by Brd4, it is unclear how P-TEFb is eventually converted to SEC that is believed to be the form of P-TEFb actually engaged in elongation. In addition, to determine how SEC contributes to transcriptional elongation in general, it is imperative to perform genome-wide analyses of the function and distribution of SEC under both normal and disease conditions. Finally, the investigation into the control of SEC formation and stability may hold the key to the development of effective strategies to reactivate latent HIV-1 and suppress the progression of aggressive acute leukemias.
References
Ammosova T, Jerebtsova M, Beullens M, Lesage B, Jackson A, Kashanchi F, Southerland W, Gordeuk VR, Bollen M, Nekhai S (2005) Nuclear targeting of protein phosphatase-1 by HIV-1 Tat protein. J Biol Chem 280:36364–36371
Ammosova T, Yedavalli VR, Niu X, Jerebtsova M, Van Eynde A, Beullens M, Bollen M, Jeang KT, Nekhai S (2011) Expression of a protein phosphatase 1 inhibitor, cdNIPP1, increases CDK9 threonine 186 phosphorylation and inhibits HIV-1 transcription. J Biol Chem 286:3798–3804
Ayton PM, Cleary ML (2003) Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev 17:2298–2307
Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM (2001) NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell 8:327–337
Barboric M, Yik JH, Czudnochowski N, Yang Z, Chen R, Contreras X, Geyer M, Matija Peterlin B, Zhou Q (2007) Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res 35:2003–2012
Biglione S, Byers SA, Price JP, Nguyen VT, Bensaude O, Price DH, Maury W (2007) Inhibition of HIV-1 replication by P-TEFb inhibitors DRB, seliciclib and flavopiridol correlates with release of free P-TEFb from the large, inactive form of the complex. Retrovirology 4:47
Bisgrove DA, Mahmoudi T, Henklein P, Verdin E (2007) Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci USA 104:13690–13695
Chao SH, Price DH (2001) Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem 276:31793–31799
Chen C, Liu M, Li H, Xue Y, Ramey WN, He N, Ai N, Luo H, Zhu Y, Zhou N, Zhou Q (2008) PP2B and PP1α cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev 22:1356–1368
Chen R, Yang Z, Zhou Q (2004) Phosphorylated positive transcription elongation factor b (P-TEFb) is tagged for inhibition through association with 7SK snRNA. J Biol Chem 279:4153–4160
Contreras X, Lenasi T, Peterlin BM (2006) HIV latency: present knowledge, future directions. Future Virol 1:733–745
Contreras X, Barboric M, Lenasi T, Peterlin BM (2007) HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog 3:1459–1469
Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J, Peterlin BM (2009) Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J Biol Chem 284:6782–6789
Core LJ, Lis JT (2008) Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science 319:1791–1792
Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL (2003) Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev 17:3029–3035
D'Orso I, Frankel AD (2010) RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat Struct Mol Biol 17:815–821
Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K (2003) The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci USA 100:8758–8763
Dey A, Ellenberg J, Farina A, Coleman AE, Maruyama T, Sciortino S, Lippincott-Schwartz J, Ozato K (2000) A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol Cell Biol 20:6537–6549
Eberhardy SR, Farnham PJ (2001) c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J Biol Chem 276:48562–48571
Estable MC, Naghavi MH, Kato H, Xiao H, Qin J, Vahlne A, Roeder RG (2002) MCEF, the newest member of the AF4 family of transcription factors involved in leukemia, is a positive transcription elongation factor-b-associated protein. J Biomed Sci 9:234–245
Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA (2007) A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130:77–88
Hargreaves DC, Horng T, Medzhitov R (2009) Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138:129–145
Harper DP, Aplan PD (2008) Chromosomal rearrangements leading to MLL gene fusions: clinical and biological aspects. Cancer Res 68:10024–10027
He N, Pezda AC, Zhou Q (2006) Modulation of a P-TEFb functional equilibrium for the global control of cell growth and differentiation. Mol Cell Biol 26:7068–7076
He N, Jahchan NS, Hong E, Li Q, Bayfield MA, Maraia RJ, Luo K, Zhou Q (2008) A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol Cell 29:588–599
He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q (2010) HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell 38:428–438
Huang F, Wagner M, Siddiqui MA (2004) Ablation of the CLP-1 gene leads to down-regulation of the HAND1 gene and abnormality of the left ventricle of the heart and fetal death. Mech Dev 121:559–572
Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K (2005) The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19:523–534
Jeanmougin F, Wurtz JM, Le Douarin B, Chambon P, Losson R (1997) The bromodomain revisited. Trends Biochem Sci 22:151–153
Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Therien C, Bergeron D, Bourassa S, Greenblatt J, Chabot B, Poirier GG, Hughes TR, Blanchette M, Price DH, Coulombe B (2007) Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell 27:262–274
Kanazawa S, Okamoto T, Peterlin BM (2000) Tat competes with CIITA for the binding to P-TEFb and blocks the expression of MHC class II genes in HIV infection. Immunity 12:61–70
Kanazawa S, Soucek L, Evan G, Okamoto T, Peterlin BM (2003) c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene 22:5707–5711
Kao SY, Calman AF, Luciw PA, Peterlin BM (1987) Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 330:489–493
Klichko V, Archin N, Kaur R, Lehrman G, Margolis D (2006) Hexamethylbisacetamide remodels the human immunodeficiency virus type 1 (HIV-1) promoter and induces Tat-independent HIV-1 expression but blunts cell activation. J Virol 80:4570–4579
Krueger BJ, Varzavand K, Cooper JJ, Price DH (2010) The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK. PLoS ONE 5(8):e12335
Kuras L, Struhl K (1999) Binding of TBP to promoters in vivo is stimulated by activators and requires Pol II holoenzyme. Nature 399:609–613
Lassen K, Han Y, Zhou Y, Siliciano J, Siliciano RF (2004) The multifactorial nature of HIV-1 latency. Trends Mol Med 10:525–531
Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A (2010) AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 37:429–437
Loyola A, Almouzni G (2004) Bromodomains in living cells participate in deciphering the histone code. Trends Cell Biol 14:279–281
Mancebo HS, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, Flores O (1997) P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev 11:2633–2644
Margolis DM (2010) Mechanisms of HIV latency: an emerging picture of complexity. Curr HIV/AIDS Rep 7:37–43
Marsden MD, Zack JA (2009) Eradication of HIV: current challenges and new directions. J Antimicrob Chemother 63:7–10
Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, Sedore SC, Price JP, Price DH, Lania L, Bensaude O (2004) Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J 23:2608–2619
Mochizuki K, Nishiyama A, Jang MK, Dey A, Ghosh A, Tamura T, Natsume H, Yao H, Ozato K (2008) The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J Biol Chem 283:9040–9048
Mueller D, Garcia-Cuellar MP, Bach C, Buhl S, Maethner E, Slany RK (2009) Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol 7:e1000249
Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL, Slany RK (2007) A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 110:4445–4454
Muniz L, Egloff S, Ughy B, Jady BE, Kiss T (2010) Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog 6:e1001152
Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, Adelman K (2007) RNA polymerase is poised for activation across the genome. Nat Genet 39:1507–1511
Natarajan M, August A, Henderson AJ (2010) Combinatorial signals from CD28 differentially regulate human immunodeficiency virus transcription in T cells. J Biol Chem 285:17338–17347
Nguyen VT, Kiss T, Michels AA, Bensaude O (2001) 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 414:322–325
Peterlin BM, Price DH (2006) Controlling the elongation phase of transcription with P-TEFb. Mol Cell 23:297–305
Ptashne M (2005) Regulation of transcription: from lambda to eukaryotes. Trends Biochem Sci 30:275–279
Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA (2010) c-Myc regulates transcriptional pause release. Cell 141:432–445
Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, Michael LH, De Mayo FJ, Schneider MD (2002) Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med 8:1310–1317
Saunders A, Core LJ, Lis JT (2006) Breaking barriers to transcription elongation. Nat Rev Mol Cell Biol 7:557–567
Schulte A, Czudnochowski N, Barboric M, Schonichen A, Blazek D, Peterlin BM, Geyer M (2005) Identification of a cyclin T-binding domain in Hexim1 and biochemical analysis of its binding competition with HIV-1 Tat. J Biol Chem 280:24968–24977
Sedore SC, Byers SA, Biglione S, Price JP, Maury WJ, Price DH (2007) Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res 35:4347–4358
Shilatifard A, Lane WS, Jackson KW, Conaway RC, Conaway JW (1996) An RNA polymerase II elongation factor encoded by the human ELL gene. Science 271:1873–1876
Shilatifard A, Duan DR, Haque D, Florence C, Schubach WH, Conaway JW, Conaway RC (1997) ELL2, a new member of an ELL family of RNA polymerase II elongation factors. Proc Natl Acad Sci USA 94:3639–3643
Slany RK (2009) The molecular biology of mixed lineage leukemia. Haematologica 94:984–993
Slany RK, Lavau C, Cleary ML (1998) The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol Cell Biol 18:122–129
Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M (2010) HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell 38:439–451
Taki T, Ohnishi H, Shinohara K, Sako M, Bessho F, Yanagisawa M, Hayashi Y (1999) AF17q25, a putative septin family gene, fuses the MLL gene in acute myeloid leukemia with t(11;17)(q23;q25). Cancer Res 59:4261–4265
Trono D, Van Lint C, Rouzioux C, Verdin E, Barre-Sinoussi F, Chun TW, Chomont N (2010) HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science 329:174–180
Tyagi M, Pearson RJ, Karn J (2010) Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol 84:6425–6437
Vlach J, Pitha PM (1993) Hexamethylene bisacetamide activates the human immunodeficiency virus type 1 provirus by an NF-kappa B-independent mechanism. J Gen Virol 74(Pt 11):2401–2408
Wei P, Garber ME, Fang SM, Fischer WH, Jones KA (1998) A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92:451–462
Xue Y, Yang Z, Chen R, Zhou Q (2010) A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res 38:360–369
Yang Z, He N, Zhou Q (2008) Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol Cell Biol 28:967–976
Yang Z, Zhu Q, Luo K, Zhou Q (2001) The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414:317–322
Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q (2005) Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 19:535–545
Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q (2003) Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell 12:971–982
Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML (2010) A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 17:198–212
Zhou Q, Yik JH (2006) The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol Mol Biol Rev 70:646–659
Zhu Y, Pe'ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price DH (1997) Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev 11:2622–2632
Acknowledgements
This work is supported by grants from the National Institutes of Health (R01AI41757-11 and R01AI41757-11 S1) to Q.Z. and a dissertation fellowship (D09-B-301) from California HIV/AIDS Research Program to N.H.
Conflicts of interest statement
The authors of this manuscript wish to state that there are no conflicts of interest involved in this work.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
He, N., Zhou, Q. New Insights into the Control of HIV-1 Transcription: When Tat Meets the 7SK snRNP and Super Elongation Complex (SEC). J Neuroimmune Pharmacol 6, 260–268 (2011). https://doi.org/10.1007/s11481-011-9267-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-011-9267-6