
Abstract
To investigate the potential of association genetics for willow breeding, Salix viminalis germplasm was assembled from UK and Swedish collections (comprising accessions from several European countries) and new samples collected from nature. A subset of the germplasm was planted at two sites (UK and Sweden), genotyped using 38 SSR markers and assessed for phenological and biomass traits. Population structure, genetic differentiation (F ST ) and quantitative trait differentiation (Q ST ) were investigated. The extent and patterns of trait adaptation were assessed by comparing F ST and Q ST parameters. Of the 505 genotyped diploid accessions, 27 % were not unique. Genetic diversity was high: 471 alleles was amplified; the mean number of alleles per locus was 13.46, mean observed heterozygosity was 0.55 and mean expected heterozygosity was 0.62. Bayesian clustering identified four subpopulations which generally corresponded to Western Russia, Western Europe, Eastern Europe and Sweden. All pairwise F ST values were highly significant (p < 0.001) with the greatest genetic differentiation detected between the Western Russian and the Western European subpopulations (F ST = 0.12), and the smallest between the Swedish and Eastern European populations (F ST = 0.04). The Swedish population also had the highest number of identical accessions, supporting the view that S. viminalis was introduced into this country and has been heavily influenced by humans. Q ST values were high for growth cessation and leaf senescence, and to some extent stem diameter, but low for bud burst time and shoot number. Overall negative clines between longitudinal coordinates and leaf senescence, bud burst and stem diameter were also found.
Similar content being viewed by others

Introduction
Plant breeding still relies, in the main, on the exploitation of existing natural variation. Biotechnology offers routes to introduce genes, and mutation breeding to induce new variants, but breeders continually search for new sources of variation among natural populations for use in crossing and selection programmes. Advances in marker technologies and quantitative statistical genetics have led to many powerful approaches for identifying gene-trait associations, even in species with complex genetic systems (Harfouche et al. 2012). Amongst these is association mapping, or linkage disequilibrium (LD) mapping, as it is also known (Khan and Korban 2012). Association mapping is considered attractive as it offers ways round the limitations of classical (linkage) genetic mapping, in which parents are crossed to form segregating families. Linkage mapping has the advantage that pedigrees and population structure are known but the disadvantage that the number of recombination events is limited. Association mapping, on the other hand, can exploit the LD and greater historical recombination available in natural populations. The tighter mapping that can be achieved increases the chances of identifying genes underlying quantitative trait loci (QTL), which can then be directly selected for (Khan and Korban 2012). Furthermore, significant associations may be of wider relevance as more alleles are surveyed than in more conventional QTL mapping studies (Hall et al. 2010), which are often based on a single bi-parental cross. Ideally, association mapping is performed using a genome wide set of markers, with the number required depending on the extent of linkage disequilibrium. For studies in systems where large number of markers are required (where linkage disequilibrium extends only a short distance), and given the need to genotype large numbers of individuals, the costs of a genome-wide approach can be prohibitive. As an alternative, a candidate gene approach can be used, where only markers positioned in genes that are thought to be involved in the trait under study are used (Hall et al. 2010). The association mapping approach has been widely applied in many trees, including poplars (Harfouche et al. 2012). However, its successful application requires a clear understanding of the genetic structure of natural populations to avoid the downstream detection of spurious associations.
Both natural evolutionary (e.g. selection, mutation, genetic drift and migration (gene flow)) and anthropogenic processes (e.g. habitat destruction, movement and displacement) influence the population structure and genetic diversity of species, as well as their distribution ranges. Depending on the relative importance of these processes, genetic and phenotypic differentiation can develop, whilst displacement may lead to establishment in new habitats, new migrations or even invasions.
An attractive way of studying processes that determine population differentiation is to use a combination of molecular and phenotypic data. This makes it possible to disentangle neutral (stochastic) from selective (adaptive) processes. For example, neutral molecular markers can give insights into the effects of gene flow, drift and to a lesser extent mutations, whilst one way of demonstrating that selection is operating is to combine individuals from different environments into one “population” which is then planted and assessed phenotypically at one or several contrasting sites. The knowledge gained is important for predicting the ability of a species, or a population, to adapt to new environments and changing climates or to new habitats if movement is required for conservation reasons. Knowledge of adaptive potential is also important for selecting accessions/populations with desired traits for use in breeding programs.
One commonly used approach for investigating the occurrence of selection is the F ST vs. Q ST comparison (Leinonen et al. 2013). The rationale of the test is that F ST estimated from molecular data provides a neutral estimate of genetic differentiation that is compared with the level of differentiation estimated from phenotypic data (Q ST ) with the aim of determining whether the phenotypic trait under study is diverging at a rate that is faster, slower, or equal to that expected under neutrality (differentiation by drift and gene flow). Thus, the method can be used to detect selection at the phenotypic trait level. Using this method, clinal variation in different morphological and life-history traits, such as growth cessation and bud burst, of various tree species have been shown to be adaptive (Savolainen et al. 2007; Ingvarsson et al. 2006). In this study we applied F ST vs. Q ST comparisons in the European willow Salix viminalis to study genetic differentiation and assist with the development of a population suitable for association mapping of traits relevant to biomass productivity.
Willows (Salix spp.) grown as short rotation coppice (SRC) are among the most important woody bioenergy crops of northern temperate regions (Hanley and Karp 2013; Karp et al. 2011). S. viminalis was identified early on as one of the species most suitable for SRC due to its fast initial growth rate, perennial habit, ability to repeatedly regrow vigorously from coppiced stools and favourable environmental credentials (Larsson 1998; Stott 1984). As a result, it has been a key species in breeding willows for bioenergy production in Europe, dominating many breeding pedigrees, and several molecular genetic mapping studies have been based on this species. Several linkage maps have been constructed with S. viminalis using both intra- and inter-specific crosses e.g. K3 and K8 used by Hanley and Karp (2013) and Hanley et al. (2002); and S1 and S3 used by Berlin et al. (2010) and Tsarouhas et al. (2002). Efforts to link phenotype to genotype have also been undertaken, through QTL mapping in these and other bi-parental populations (Tsarouhas et al. 2002, 2003, 2004; Semerikov et al. 2003; Rönnberg-Wästljung et al. 2008; Samils et al. 2011; Höglund et al. 2012). However, to date, association mapping has not been applied in this species.
S. viminalis is a pioneer shrub of riparian environments and is widely distributed across Europe, from the Atlantic in the west to Central Siberia in the east, and at latitudes from Scandinavia in the north to the Mediterranean Sea in the south. It is dioecious, insect and wind pollinated (Alström-Rapaport 1997) and propagates sexually by seeds that are dispersed by wind or water, and vegetatively, through the rooting of broken twigs and branches. In addition, humans have undoubtedly influenced S. viminalis through a long history of cultivation, principally for basketry and weaving and also for management of river banks and waterways where this species is often found naturally (Kuzovkina et al. 2008). The degree of human influences on the distribution of S. viminalis across Europe is not fully known however, and may be country-specific. In England, for example, where many species used for basketry are native, the cultivation of willows is specifically mentioned by the Roman historian Pliny the Elder and wickerwork fragments were unearthed from the UK Glastonbury Lake Village dated 100 BC (Karp 2013). In Sweden, however, S. viminalis is not native but was first introduced in the seventeenth century, followed by several more recent introductions (Lascoux et al. 1996). This transfer of basket willows, often across widely separated geographic regions, is in contrast to more recent spreading of individual willows naturally by broken branches floating along streams. The latter can often be distinguished by the occurrence of individuals downstream in the locality carrying identical genotypes to those found further up (Beismann et al. 1997). Clearly, efforts aimed at using association mapping approaches in S. viminalis sampled from natural populations need to assess and take into account these potential factors.
Accessions of S. viminalis collected from nature already exist in some European collections. As a first step towards the application of association mapping in willow, we have combined the S. viminalis collections in Sweden and the UK and augmented these with S. viminalis sourced from natural populations in Europe. After initial molecular marker analysis, a combined subset of accessions was planted in fully replicated field trials at contrasting sites in Sweden and the UK. The accessions were assessed for multiple traits, over multiple years, to provide information of the phenotypic variation represented.
To investigate factors and processes shaping the population structure and distribution of genetic variation in the subset of accessions, the data were used to estimate quantitative trait variation in the form of Q ST . The germplasm was genotyped using 38 SSR markers, and the data were subsequently used to assess genetic structure and differentiation (F ST ) across subpopulations. The extent and patterns of trait adaptation were assessed by comparing population genetic (F ST ) and quantitative genetic (Q ST ) parameters. The results are discussed in the context of population structure, environmental conditions and migration history of S. viminalis in Europe.
Material and methods
Plant material
To establish a diverse collection of S. viminalis accessions, germplasm from multiple European collections were brought together. The largest collections were the UK National Willow Collection at Rothamsted Research (Harpenden, UK) and the Swedish University of Agricultural Sciences collection (Uppsala, Sweden). These collections represent genotypes that originated from the UK, Sweden, Belgium Germany, Russia and a substantial number of accessions that were recently collected in the Czech Republic (Trybush et al. 2012). Based on the result of subsequent analyses (described later), these became grouped into four subpopulations: Eastern Europe (from the Danube and eastwards), Western Europe (from the Rhine), Western Russia or Sweden.
Some additional managed germplasm was kindly provided by the Willow for Wales collection at the Institute of Biological, Environmental and Rural Sciences (IBERS), Aberystwyth, Wales, and from the willow collection at the Forestry and Game Management Research Institute in Kunovice, Czech Republic. Additional material was collected from unmanaged willow stands near to Radojewo and Kazimierz Dolny in Poland and from several locations in Scania, southern Sweden. Similarly, additional samples were collected from the banks of the rivers Wye, Severn and Teme in the UK. These UK samples were only included in microsatellite analyses and were not planted in the field trials due to their later acquisition. Samples originated from latitudes between 48.1°N to 62.4°N and longitudes from 4.8°W to 104.3°E. The final number of accessions available to this study was 512, of which 508 were successfully genotyped (Supplementary Table 1).
Field experiments
In spring 2009, field experiments were established at two different sites with 20 cm cuttings from selected genotypes. One site was Pustnäs, south of Uppsala, Sweden (59.80°N, 17.67°E, 25 m Above Ordinance Datum (AOD)) and the other was at Woburn, UK (52.01°N, 0.59°W; 95 m AOD). The Pustnäs site is a sandy loam, while Woburn is a sandy loam of coarse texture (>70 % sand, 63–2,000 μm) to at least 0.7 m depth. A total of 387 accessions per block in Pustnäs and 397 at Woburn were planted as single plants in six blocks according to a randomized complete block design. The different number of accessions planted at each site resulted from the limited availability of some cuttings for planting. The spacing was 130 cm between rows and 50 cm between plants within rows. Two border rows (genotype 78183) were planted outside the experimental plants to reduce border effects. During the first growing season, no fertiliser was applied to the experiments; but during years 2010 and 2011, the experiments were fertilised with N P K (21-4-7) corresponding to 80 kg N ha−1 year−1. Plants were cut back to the stool in winter 2009/2010.
Phenotypic measurements
Leaf bud burst time was assessed on each individual plant in both field experiments during spring 2010 (BB10) and 2011 (BB11), and in Pustnäs in 2013 (BB13) using a scale developed by Weih (2009) where 1 = no sign of bud growth, tip of the bud tightly pressed to the shoot; 2 = bud swollen, tip of the bud bending from the stem, shoot tip 1–4 mm long; 3 = green foliage showing, shoot tip longer that 4 mm; 4 = elongating, new leaves starting to disentangle from each other; 5 = one or more leaves are perpendicular to the shoot axis. In Pustnäs, the bud burst assessments were done repeatedly every second day during a 2-week period in order to include the time point where most variation in bud burst was seen. In Woburn, one specific time point was chosen. Growth cessation, defined by shoot apex abscission, was assessed in Pustnäs in autumn 2010 (GC10) on the highest shoot of each plant. The measurements were done from the end of August and once or twice a week depending on the rate of progression. The plants were scored for the response according to the day they had reached growth cessation. So, plants that had reached growth cessation by the first day of measurement were given a value of 1, and so on. Leaf senescence and abscission were visually estimated in Pustnäs in autumn 2010 (LS10) and 2011 (LS11), and in Woburn in 2010 (LS10) according to a leaf senescence index (LSI) from 0 to 4 where 0 = no leaves left on the plant (100 % abscission); 0.5 = less than 10 % brownish leaves (~95 % abscission); 1 = 10 to 20 % brownish leaves (~85 % abscission); 1.5 = 20 to 30 % brownish or yellow leaves (~75 % abscission); 2 = 30 to 40 % yellow leaves (~65 % abscission); 2.5 = 40 to 50 % yellow and green leaves (~55 % abscission); 3 = 50 to 65 % green leaves (~40 % abscission); 3.5 = 65 to 80 % green leaves (~30 % abscission) and 4 = more than 80 % green leaves (~10 % abscission) (Ghelardini et al. 2014). Two biomass traits were assessed during winter 2010/2011 at both sites. The number of shoots (Nsh11) was counted and diameters on all shoots above 55 cm from the ground (Sdia11) were measured and summed. Where included in the field trials, plants that were initially thought to be of a unique genotype but later assigned to an identical accession group were treated as a single genotype, with phenotypic data from all planted occurrences of the genotype used as biological replicates.
Molecular genetic analyses
Genomic DNA from the 508 available accessions was isolated from fresh leaf tissue using the DNeasy Mini Plant Kit (Qiagen, Crawley, UK). Screening of 38 SSR loci was conducted as described in Trybush et al. (2012). Marker details are provided in Supplementary Table 2. Based on the marker data, three accessions appeared to be polyploids and were removed from further analyses. First, using all 38 markers, clusters were identified using STRUCTURE v 2.3.2.1 (Pritchard et al. 2000; Falush et al. 2003). An admixture model was employed where correlated allele frequencies were assumed, and the K value (i.e. the number of clusters) was set from one to eight, a value within this range expected to be the most likely result. The length of the burn-in period was set to 10,000, and the Monte Carlo Markov Chain (MCMC) model after burn-in was run for an additional 100,000 iterations. For each K, 40 replicates were run and runs with an outlier value of lnPD were removed (χ 2 test, α = 0.05, as implemented in R-package ‘outliers’). The optimal value of K was determined by examination of the L(K) and Evanno’s ∆K statistics (Evanno et al. 2005) using the R package ‘corrsieve’. Then, in each cluster, null alleles were identified using the Cervus 3.0 software (Kalinowski et al. 2007). Three loci (SB1366, SB355 and SB1464) that contained null alleles with high probabilities were removed, leaving 35 loci that were used in all subsequent molecular genetic analyses. Based on the marker data, groups of accessions with putatively identical genotypes were also identified. In subsequent molecular analyses, one accession per group was used to ensure that only one representative of each multi-locus genotype was analysed, resulting in 369 unique accessions. Using that dataset, population structure was again investigated using STRUCTURE v 2.3.2.1 (Pritchard et al. 2000; Falush et al. 2003), with the same parameter settings as described above. Posterior probability of accessions belonging to a specific cluster for which the exact origin was known were superimposed on a map using ArcView 9.3.1 (ESRI 2011). Based on the results from this analysis, each accession was assigned to one of four subpopulations Eastern Europe, Western Europe Western Russia or Sweden.
The distances between the identical accessions within the groups (for which sampling coordinates were available) were compared among as well as within each of the four subpopulations. The distances in sampling latitudinal coordinates (in degrees) were compared between the identical accessions in each group. The groups were then classified in one of three categories (A, B or C), depending on the distances between the sampling coordinates of the identical accessions within the groups. The categories were A = <1° apart, B = 1–5° apart or C = > 5° apart.
Analyses of markers
GenAIEx v 6.5 (Peakall and Smouse 2012) was used to determine the sample size, number of alleles, number of effective alleles, observed heterozygosity and expected heterozygosity for each of the 35 markers. Departure from Hardy–Weinberg equilibrium was also calculated for each marker using GenAIEx v 6.5
Isolation by distance
The genetic isolation by distance was investigated by correlating the genetic distance between samples with the geographic distance. Only accessions for which sampling coordinates were available were used and identical accessions that were sampled in more than one subpopulation were also excluded (N = 285). Rousset’s distance (Rousset 2000) between pairs of accessions (a, analogous to the FST/(1-FST) ratio) were calculated using SPAGeDi v 1.3. (Hardy and Vekemans 2002). Distances between accessions were calculated using the Perl module GIS::Distance::Formula::Vincenty. The resolution of sampling location was limited for some of the accessions, resulting in a distance of zero for some pairs. Such pairwise distances were therefore replaced by a random short distance between 1 and 500 m. The significance of the correlations was assessed by Mantel test, using R library ‘ade4’.
F ST statistics and genetic diversity in subpopulations
F ST values were estimated using Arlequin v 3.5 (Excoffier and Lischer 2010). Based on the results of molecular genetic analyses, each accession was assigned to one of the four subpopulations: Eastern Europe (from the Danube and eastwards), Western Europe (from the Rhine), Western Russia and Sweden. Only accessions that were present in both field experiments and for which sampling coordinates were known were included (N = 258). Sample size, number of alleles, number of effective alleles, observed heterozygosity and expected heterozygosity were calculated in each of these subpopulations using GenAlEx v 6.5 (Peakall and Smouse 2012). Departure from Hardy-Weinberg equilibrium was also calculated for each marker in each subpopulation using GenAIEx v 6.5.
Quantitative genetic analyses
In order to obtain accession predictors (best linear unbiased estimates, BLUE) along with individual predictors for each plot adjusted for spatial variation for further analysis, linear mixed models were fitted to the phenotypic data. For this purpose a Residual Maximum Likelihood (REML) method implemented in GenStat (2011, 14th edition, © VSN International Ltd, Hemel Hempstead, UK) was used. No transformation was required for the bud burst data from either site. A square root transformation was used for growth cessation data for Pustnäs (GC10) and for Woburn leaf senescence (LS10) to ensure a Normal distribution and homogeneity of variance. This transformation was also used for number of shoots (Nsh11) data for Pustnäs and for the sum of diameters (Sdia11) data from both sites. Models were of the form:
where Y iklm is the (transformed where required) phenotypic value in the ith block for the kth accession located at row l and column m, with μ the overall mean, B i the fixed effect of the ith block, C k the fixed effect of the kth accession, (Row.Col) lm the interaction effect of row l and column m and e iklm the residual error. Where required in terms of significant (p < 0.05) change in the χ 2-distributed model deviance, first order autocorrelation terms were imposed (Cullis and Gleeson 1991) on rows and columns in the models. This was required in all the models of Pustnäs data but for Woburn, such a term was only required for columns for bud burst data in 2010 (BB10). For the traits analysed (Table 4), the average percentage change of a raw plot (single plant) value to a spatially corrected one for Pustnäs was 9.4 %, (BB10), 11.2 % (BB11), 11.7 % (LS10), 16.19 % (Nsh11) and 16.46 % (Sdia11); for Woburn, it was 2.22 % (BB10).
In order to partition the quantitative trait variation between and within subpopulations and to estimate the Q ST , the individual predictors adjusted for spatial variation (Y pr = μ + B + C + e in Eq. 1) were used for modelling. The traits BB10, BB11, BB13, GC10, LS10, LS11, Nsh11 and Sdia11 at both sites (Pustnäs and Woburn) were analysed, but the analysis was restricted to the same set of genotypes (N = 258) used in the F ST analysis described above. Variance components were estimated using REML in the analysis of variance module in JMP ® v 8 with the linear mixed model:
where P j is the random effect of the jth population, C(P) jk the random effect of the kth accession hierarchically nested within the jth population and the other model terms are the same as described in Eq. 1. An approximate Q ST estimate (Spitze 1993) assuming only additive genetic variance was estimated using the formula:
where σ2 P and σ2 CP represent the variance component between subpopulations and genotype variation within subpopulation, respectively. Q ST estimation errors were calculated using the Delta method based on the Taylor series approximation (Casella and Berger 2002). Pairwise Q ST values were estimated for each field experiment using the same approach as above. Arithmetic mean values and standard errors were reported for each trait and population. For the transformed variables the back transformed data was used for the mean calculations.
Correlations between sampling coordinates and trait values
In order to investigate whether leaf senescence 2010, bud burst 2011 and summed stem diameter 2011 at both sites showed clinal variation over latitudes and longitudes, correlations between the accession trait predictors (μ + C in Eq. 1) for each trait and sampling latitude and longitude coordinates were examined by linear regression analyses. These analyses were performed with all data (all accessions and subpopulations) as well as for each subpopulation separately. Sampling latitude and longitude coordinates were used as independent variables in addition to an intercept. The dataset was the same as for the F ST and Q ST analysis but without the groups where identical accessions were collected more than 1° from each other (N = 239). Regression analyses and t-tests of coefficient estimates were performed using the lm-function in R taking into account eventual multicollinearity.
Results
Genetic diversity and occurrence of identical accessions
Among the 505 genotyped diploid accessions, 320 had unique genotypes and 185 had a genotype that was identical to at least one other accession. Of these 185 accessions, 49 groups of unique genotypes could be identified, giving a total of 369 (73 %) unique genotypes (320 + 49; Supplementary Table 1). Within the 49 groups, the number of identical accessions ranged from two to 21, with most groups represented by few identical accessions, i.e. 41 groups contained 5 or less and 27 groups consisted of just two identical accessions. Thirty groups were classified based on the distances of identical genotypes (Table 1) of which 17 were category A (<1° apart), 10 were category B (1–5° apart) and three category C (>5° apart). In Eastern Europe all seven groups were category A, in Western Russia the single group was assigned to category C, in Sweden where the majority of the groups were found, 6 were category A and 10 were category B, and in Western Europe the four groups were all category A (Table 1). Two groups contained identical accessions in several subpopulations (Table 1).
A total of 471 alleles were amplified across the accessions. The number of alleles per locus across all accessions ranged from two to 42, with a mean of 13.46, and the number of effective alleles ranged from 1.00 to 9.44, with a mean of 3.73 (Table 2). Mean observed heterozygosity across the markers was 0.55 (ranged between 0 and 0.86) and mean expected heterozygosity was 0.62 (ranged between 0.01 and 0.89). All but one marker showed significant (p < 0.05, χ 2 test) deviations from the Hardy–Weinberg equilibrium, however there were no marked differences between observed and expected heterozygosities.
The number of genotyped accessions ranged from 22 in the Western Russian subpopulation to 134 in the Eastern European subpopulation (Table 2). The mean number of alleles across all accessions in each subpopulation ranged from 6.20 in Western Europe to 9.34 in Western Russia and the number of effective alleles ranged from 3.10 in Sweden to 5.16 in Western Russia. Mean observed heterozygosity was highest in the Western Russian subpopulation (H O = 0.60), while H O was estimated to around 0.55 in the other three subpopulations. Mean expected heterozygosity ranged from 0.57 in Sweden to 0.69 in Western Russia. There were no marked differences between observed and expected heterozygosities within the subpopulations, thus no markers warranted exclusion.
Population differentiation (F ST statistics)
Overall genetic differentiation among all analysed accessions was highly significant with a mean overall F ST of 0.06 (p < 0.0001, AMOVA). All pairwise F ST values (Table 3) were also highly significant (p < 0.001, AMOVA). with the greatest genetic differentiation detected between the Western Russian and the Western European subpopulations (F ST = 0.119). In contrast, the Swedish and the Eastern European subpopulations showed the smallest pairwise F ST (0.04).
Population structure
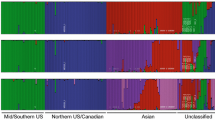
Bayesian clustering implemented within the STRUCTURE v 2.3.2.1 software identified K = 4 as the most likely number of subpopulations and this result was in accordance with both L(K) and Evanno’s ∆K estimates (Fig. 1). The four subpopulations more or less overlapped with samples originating from Western Russia, Western Europe, Eastern Europe and Sweden (Fig. 2).

Population structure of the S. viminalis accessions, clustered into K clusters using STRUCTURE v 2.3.2.1. The number of clusters K is shown for K = 2 to K = 5. Each accession is represented by a vertical bar; each bar being one of K colours representing K clusters (1st red, 2nd blue, 3rd green, 4th pink, 5th orange). The length of the coloured bar shows the accessions’ estimated proportion of membership to each cluster. The accessions are arranged according to known origin or ‘Unknown’ in cases where the origin was unknown or uncertain

Map showing the origin of a subset of the S. viminalis accessions, for which the origin was known. The symbol for each accession is a pie chart with four colours (1st red, 2nd blue, 3rd green, 4th pink), representing the four clusters, showing the estimated proportion of membership to each cluster at all sampled sites. Arrows indicate the positions of the field trials in Pustnäs, Sweden and Woburn, UK
Isolation by distance
Plotting of Rousset’s pairwise genetic distance measure a (analogous to F ST /(1 − F ST )) vs. the logarithm of the distance revealed a pattern of significant isolation by distance in all subpopulations studied except within Sweden (within Sweden p = 0.5, all other p < 0.001, Mantel test; Fig. 3). Even where it was significant, the effect in terms of regression slope was however not large (in the range 0–0.02, SE 0.00027–0.0016), and decreased as the extent of geographic distance decreased, implying that wide geographic distances are needed in order to have barriers to gene flow.

Rousset’s distance, a, analogous to pairwise F ST vs. pairwise distance, for each pair of accessions in all populations. The best linear regression fit is shown as a line. The correlations were significant (p < 0.05, F test) for all populations except Sweden
Quantitative variation and F ST vs. Q ST comparisons
Plants at Woburn had on average a substantially larger summed stem diameter and produced on average more shoots than the plants at Pustnäs (Table 4). When comparing the four subpopulations, the Western Russian subpopulation was considerably different from the others, particularly regarding leaf senescence and growth cessation times, which occurred significantly earlier in this population. This pattern was consistent across years and sites. Also the summed stem diameters were significantly smaller in the Western Russian subpopulation and there was a tendency for the total number of shoots to be fewer as well. In addition, bud burst was somewhat earlier in the Western Russian and East European compared with the other two subpopulations. This tendency was most prominent for the Western Russian subpopulation in Woburn 2011. As a consequence, the overall Q ST values across all subpopulations were highest for growth cessation and leaf senescence and rather high for summed stem diameter. Number of shoots and bud burst (except for Woburn 2011) had low Q ST values (Table 4). All pairwise F ST vs. Q ST comparisons with Western Russia for leaf senescence and growth cessation had higher Q ST than F ST values (Fig. 4). In all other cases there were no strong signals of higher Q ST than F ST values, except for bud burst 2011 in Woburn where the comparisons with the Western Russian population had higher Q ST compared to F ST .

F ST and Q ST between all population pairs. Q ST values from Woburn are shown in the top graph; Q ST values from Pustnäs are shown in the lower graph. Error bars represent 95 % CI for F ST and SE for the Q ST . For each trait, from left to right, the population pairs compared are: East Europe vs. West Russia, East Europe vs. Sweden, East Europe vs. West Europe, West Russia vs. Sweden, West Russia vs. West Europe and Sweden vs. West Europe. Codes for traits are: BB—bud burst, GC—growth cessation, LS—leaf senescence, Nsh—no. of shoots, Sdia—summed stem diameter, followed by the year, 10 (2010) or 11 (2011) or 13 (2013)
Correlations between sampling coordinates and trait values
There were significant (p < 0.01, t tests) longitudinal clines in all examined traits when including all data and this pattern was seen at both Pustnäs and Woburn (Fig. 5a). Overall slope coefficients were negative for leaf senescence indicating earlier leaf senescence with increasing longitude. The overall coefficients were also negative for sum of stem diameters (smaller diameter with increasing longitude) whereas they were positive for bud burst (earlier bud burst with increasing longitude). Strikingly, overall longitudinal clines were considerably influenced by the Western Russian subpopulation. However, even without the Western Russian subpopulation, the longitudinal slope coefficient directions were the same as with all data for budburst and leaf senescence, but not for stem diameter. For leaf senescence in Woburn and for budburst at both sites the coefficients were still strongly significant (p < 0.001, t tests). When analysing each subpopulation separately, only BB11 in the Eastern European subpopulation in Pustnäs had a slope significantly (p < 0.05, t test) different from zero.


Scatterplot of accession predictors for leaf senescence 2010, budburst 2011 and summed stem diameter 2011 against accession origin longitude (a) and latitude (b) for each of the two sites (Pustnäs and Woburn). Latitude/longitude regression lines modelled using all accessions are shown in black, while accessions and regression lines originating from Sweden, West Russia, East Europe and West Europe are depicted in red, pink, green and blue, respectively. Significance of regression slopes are indicated with *(p < 0.05), **(p < 0.01) and ***(p < 0.001). Data for summed stem diameter and leaf senescence in Woburn were transformed by square root prior to analysis
When analysing all data, there were also significant (p < 0.001, t tests) latitudinal clines for leaf senescence with negative slope coefficients (earlier leaf senescence with increasing latitude) at both sites, and when examining each subpopulation separately this pattern was found in all subpopulations except for Sweden (Fig. 5b). There was also an overall latitudinal cline for bud burst with negative slope coefficients (later bud burst with increasing latitude) albeit significant (p < 0.001, t test) only at Pustnäs. No significant (p < 0.05, t test) latitudinal gradient was found for summed stem diameter.
It should be noted that the proportions of the accession variation that were predicted using these linear regressions models (R 2) were rather limited. The R 2 estimates for leaf senescence were the highest (in the range 0.45–0.70), while the corresponding estimates for budburst and summed diameter were lower (0.03–0.32 and 0.01–0.08), respectively.
Discussion
S. viminalis is native to many European countries. It has historically been utilised and cultivated for basket-making, hurdles and riverbank stabilisation, and is still used in these small industries. However, grown as SRC, it has also become important as a biomass crop for bioenergy production. It has been widely used in breeding programmes and many genetic maps have been constructed and used in QTL analysis but, until now, association mapping has not been applied. In this study, we report on the genetic diversity and population structure of a population of S. viminalis collected from different parts of Europe for association mapping. Significant differentiation and structure were revealed. These findings will need to be taken into account when using the population for association genetics but they have also shed light on the factors shaping the genetic structure of this widespread European willow.
Identical accessions
Our analyses revealed that a large number of accessions (27 %) among the sampled plants had identical genotypes for all 35 markers and could therefore be considered as clones. This is not surprising as willows reproduce vegetatively through the rooting of branches in addition to reproducing sexually as seeds through obligate outcrossing of dioecious parents. Vegetative propagation via cuttings has also been exploited by humans as a means of cloning and transplanting genotypes with favourable characteristics.
In nature, the relative importance of asexual vs. sexual reproduction among Salicaceae appears to differ greatly (Karrenberg et al. 2002) but recent studies suggest that successful seed dispersal and germination may be more common than previously thought, at least in certain environments (Trybush et al. 2012). It is also known that vegetative propagation, resulting from broken branches that float in the water and root when appropriate conditions are met, is a common occurrence. Thus, identical genotypes can often be found some distance apart along riverbanks and quite far downstream of putative ‘parental’ genotypes, although sexual reproduction may still be occurring and populations can be a mixture of sexual and vegetative offspring (Beismann et al. 1997; Shafroth et al. 1994). A high degree of vegetative propagation also seems to occur when certain species are introduced into new areas, e.g. Salix purpurea introduced to the USA compared to the native Salix eriocephala (Lin et al. 2009).
In the present study, we postulated that natural propagation would most likely result in identical individuals being found relatively close to each other, and/or in a location relative to possible donor genotypes upstream along a river course, whilst artificial propagation by humans would result in identical genotypes being found further apart in geographically unrelated areas. According to this rationale, we grouped identical accessions on the basis of distance and found that 17 contained identical accessions that were close to each other (category A), while 13 groups contained identical accessions at larger distances apart (category B and C). This suggests that vegetative propagation in S. viminalis is the result of both natural and artificial vegetative propagation. In the latter case, the most likely explanation is that humans have propagated cuttings for the basket industry, for the production of windbreaks, for stream/river bank stabilization or, more recently, for use as a biomass crop. Our results are similar to those described in the previous studies of Salix spp. (Beismann et al. 1997; Shafroth et al. 1994; Lin et al. 2009; Trybush et al. 2012) and to studies of Populus nigra in Moravia, Czech Republic (Pospísková and Bartáková 2004), the UK (Winfield et al. 1998) and Europe (Smulders et al. 2008).
Genetic diversity, population differentiation and structure
All standard population genetic measures used (e.g. mean number of alleles per locus 13.46, mean H O 0.55, mean H E 0.62) indicated that overall levels of genetic diversity were similar to those reported in an earlier study based on a subset of the S. viminalis accessions used here that were collected in the Czech Republic and analysed with the same panel of SSR markers (mean number of alleles per locus 6.95, mean H O 0.55, mean H E 0.56, respectively; Trybush et al. 2012). The results were also similar to those reported for populations of Salix exigua (Douhovnikoff et al. 2005) and Salix herbacea (Alsos et al. 2009) assessed using Amplified Fragment Length Polymorphisms (AFLPs).
Based on the analyses of 35 microsatellite loci, we found evidence for well-defined population structure. Four well-separated clusters were revealed corresponding to samples originating from: Sweden, Western Europe, Eastern Europe and Western Russia.
The overall F ST value (0.06) indicated the presence of statistically significant differentiation, as has also been reported previously for S. viminalis (Trybush et al. 2012) as well as for S. herbacea (Alsos et al. 2009). In the present study, the Western Russian subpopulation was the most differentiated, with high F ST values in all pairwise comparisons. This subpopulation also had a slightly higher mean observed heterozygosity (H O = 0.60) and more alleles than the other subpopulations (H O = 0.55), even though fewer samples were analysed. This could mean that the Western Russian subpopulation has a higher effective population size than the other subpopulations.
The lowest pairwise F ST value was between the Swedish and the Eastern European subpopulations. The majority of the groups containing identical accessions in the present study were also found in the Swedish subpopulation. These were also all category B accessions, suggesting high levels of anthropogenic vegetative propagation. These results support the view that S. viminalis was introduced into Sweden and concurs with the results from Lascoux et al. (1996), who found that the Swedish S. viminalis population was different from Central European populations and comprised individuals with a mixed background. Further support for the Swedish subpopulation being a mixture of introduced clones is the lack of isolation by distance in this subpopulation compared to the others, for which statistically significant isolation by distance was detected, as well as a consistent lack of clinal variation for leaf senescence time in Sweden.
The low levels of differentiation found here between the Swedish and the Eastern European subpopulations also agrees with Lascoux et al. (1996) who concluded that the origin of the Swedish samples in their study was north-western Poland. However the clustering analysis conducted here clearly separated the Swedish subpopulation from the others, and the pairwise F ST was statistically significantly different from 0, suggesting that the Swedish population has started to differentiate from its ancestors. An alternative explanation is that we have not sampled from the precise centre of origin of the Swedish samples, which is possible as sampling was limited in north-western Poland.
In this study, an attempt was made to assemble a population of more than 500 individuals for association mapping. It is not possible to know in advance whether a given population size is adequate but the target size chosen here fits within the range successfully used in previous studies in trees (e.g. Ingvarsson et al. 2006). The occurrence of identical genotypes reduced the numbers of individuals that could be used. The fact that significant structure was identified does not negate the use of the population for association mapping but it does require that subsequent analysis takes the structure into account to reduce the possibility of false positives (spurious associations). False positives can arise where there is linkage between causal and non-causal sites, which may occur in some subpopulations. They would not be removed by increasing the total population size, or number of markers, although the ability to detect population structure is affected by marker density, population size, the level of admixture and the divergence in allele frequency between subpopulations (Larsson et al. 2013). Future use of this population for association mapping will need to account for population structure using approaches such as unified mixed linear modelling, as recent results in maize have shown that these perform best at removing false associations because they account for both population structure and relatedness between individuals (Larsson et al. 2013). The identification of subpopulations that correspond with specific regions provides some informative knowledge on gene pools for breeding programmes and justifies further collections from wider geographic regions being made to further improve our understanding of S.viminalis in Europe.
Quantitative variation and F ST vs. Q ST comparisons
High overall Q ST values were observed for growth cessation and leaf senescence times, and to a lesser extent summed stem diameter, but low Q ST values were found for bud burst time and number of shoots. The high Q ST values for growth cessation, leaf senescence and summed stem diameter were mainly attributable to the West Russian subpopulation, which was highly differentiated compared to the others, initiated growth cessation earlier in the autumn and had smaller summed stem diameters. These Western Russian accessions are probably adapted to a pronounced continental climate with sharp seasonal changes (e.g. a short spring and autumn). In the conditions at Pustnäs and Woburn, their early growth cessation means that part of the growing season is missed, which is the probable cause of their lower summed stem diameters (and thus biomass yields). Interestingly, in all pairwise comparisons with Western Russia, Q ST was higher than F ST for both growth cessation and leaf senescence, implying that natural selection is operating. A recent study of 24 willow and poplar species in a glasshouse experiment under two treatments of temperate and sub-tropical climate has suggested that environmental cues that trigger growth may be an important adaptive trait leading to trade-offs between freezing tolerance and growth rate that influence the geographical ranges of willow and poplar species (Savage and Cavender-Bares 2013).
The trait variation across the subpopulations was also evident from the overall negative longitudinal genetic clines for leaf senescence and stem diameter, and positive longitudinal clines for bud burst. These trends were again mostly attributable to the Western Russian subpopulation that was located far east of the other subpopulations. However, when the West Russian subpopulation was excluded, the pattern remained for leaf senescence and bud burst but not for stem diameter. We also found a weak overall latitudinal cline for leaf senescence (negative slope), as is expected given that the growing season is shorter closer to the poles. This pattern was found in all subpopulations except for the Swedish (where the slope was positive). There was also an overall latitudinal cline for bud burst at Pustnäs only; however, this trend could not be seen in the individual populations. There was no cline for summed stem diameter. We cannot conclude that the latitudinal clines we observed in S. viminalis resulted from natural selection since we did not observe pairwise Q ST estimates between subpopulations from different latitudes (e.g. Sweden and East Europe) to be significantly greater than the corresponding pairwise F ST estimates. In most of the cases, it is therefore more likely that the observed patterns were caused by non-adaptive processes.
Examples of clinal variation with latitude have been described for budburst in Quercus petraea (Ducousso et al. 1996), growth cessation in Betula pendula (Viherä-Aarnio et al. 2005), bud set in Pinus sylvestris (Mikola 1982) and growth cessation in Picea abies (Hannerz and Westin 2000). An example of a cline in willows linked with geographical coordinates has also recently been described in Salix reticulata, where leaf traits were assessed for eight populations from across the species range in Europe and compared with one population in the Rocky Mountains (Marcysiak 2012). Leaf size traits in Europe were correlated with increasing latitude and longitude but the correlations diminished with increasing altitude.
The F ST − Q ST differences and latitudinal/longitudinal clines observed in this study constitute a valuable resource for facilitating the selection of breeding material that is suitably adapted to different climatic conditions given a clearly formulated breeding objective. However, in order to formulate practical recommendations that would be useful in breeding, further research of this kind is required for other important traits such as plant survival, fertility/fecundity traits, and on resistance to insects and pathogens.
Overall, biomass production was higher at the Woburn site compared to the Pustnäs site. Although a combination of several factors could explain this, including soil conditions and precipitation, it is likely that the shorter growing season, and possibly the more continental climate at the more northern Pustnäs site were major contributors.
Concluding remarks
This study provides clear evidence of structure in the European S. viminalis accessions from collections in the UK and Sweden and samples from natural populations. To avoid false positives this must be taken into account when the samples are used in association mapping, genome-wide association studies, and future genomic selection approaches, for example by using analyses based on unified mixed linear models. The results also revealed high numbers of identical genotypes, particularly in Sweden, supporting the view that willows have been artificially introduced into this country by humans. Given the population substructure associated with different European regions, it would be worthwhile to make further collections and expand the geographic coverage as this may uncover new sources of diversity for breeding. Such extended sampling should, however, be structured in a way as to minimise the possibility of collecting vegetatively propagated clones. The distribution of phenotypic variation and possible signs of ongoing natural selection, particularly for leaf senescence and growth cessation traits, and the geographical clines reported here, could be taken into consideration when selecting breeding material for different climatic zones.
References
Alsos IG, Alm T, Normand S, Brochmann C (2009) Past and future range shifts and loss of diversity in dwarf willow (Salix herbacea L.) inferred from genetics, fossils and modelling. Global Ecol Biogeogr 18:223–239
Alström-Rapaport C (1997) On the sex determination and evolution of mating system in plants. Swedish University of Agricultural Sciences, Uppsala
Beismann H, Barker JHA, Karp A, Speck T (1997) AFLP analysis sheds light on distribution of two Salix species and their hybrid along a natural gradient. Mol Ecol 6:989–993
Berlin S, Lagercrantz U, von Arnold S, Öst T, Rönnberg-Wästljung AC (2010) High-density linkage mapping and evolution of paralogs and orthologs in Salix and Populus. BMC Genomics 11:129
Casella G, Berger RL (2002) Statistical inference. Duxbury Press, Belmont
Cullis BR, Gleeson AC (1991) Spatial analysis of field experiments - an extension to two dimensions. Biometrics 47:1449–1460
Douhovnikoff V, McBride JR, Dodd RS (2005) Salix exigua clonal growth and population dynamics in relation to disturbance regime variation. Ecology 86:446–452
Ducousso A, Guyon JP, Kremer A (1996) Latitudinal and altitudinal variation of bud burst in western populations of sessile oak (Quercus petraea (Matt) Liebl). Ann Sci For 53:775–782
ESRI (2011) ArcGIS Desktop: Release 10. Environmental Systems Research Institute, Redlands
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Ghelardini L, Berlin S, Weih M, Lagercrantz U, Gyllenstrand N, Rönnberg-Wästljung AC (2014) Genetic architecture of spring and autumn phenology in Salix. BMC Plant Biol 14:31
Hall D, Tegstrom C, Ingvarsson P (2010) Using association mapping to dissect the genetic basis of complex traits in plants. Brief Funct Genomics 9:157–165
Hanley SJ, Karp A (2013) Genetic strategies for dissecting complex traits in biomass willows (Salix spp.). Tree Physiol. Tree Physiology doi: 10.1093/treephys/tpt089
Hanley S, Barker A, Van Ooijen W, Aldam C, Harris L, Åhman I, Larsson S, Karp A (2002) A genetic linkage map of willow (Salix viminalis) based on AFLP and microsatellite markers. Theor Appl Genet 105:1087–1096
Hannerz M, Westin J (2000) Growth cessation and autumn-frost hardiness in one-year-old Picea abies progenies from seed orchards and natural stands. Scand J For Res 15:309–317
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Harfouche A, Meilan R, Kirst M, Morgante M, Boerjan W, Sabatti M, Mugnozza GS (2012) Accelerating the domestication of forest trees in a changing world. Trends Plant Sci 17:64–72
Höglund S, Rönnberg-Wästljung AC, Lagercrantz U, Larsson S (2012) A rare major plant QTL determines non-responsiveness to a gall-forming insect in willow. Tree Genet Genomes 8:1051–1060
Ingvarsson PK, Garcia MV, Hall D, Luquez V, Jansson S (2006) Clinal variation in phyB2, a candidate gene for day-length-induced growth cessation and bud set, across a latitudinal gradient in European aspen (Populus tremula). Genetics 172:1845–1853
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Karp A (2013) Willows as a source of renewable fuels and diverse products. In: Fenning T (ed) Challenges and opportunities for the world’s forests in the 21st century. Springer, Dordrecht
Karp A, Hanley SJ, Trybush SO, Macalpine W, Pei M, Shield I (2011) Genetic improvement of willow for bioenergy and biofuels. J Integr Plant Biol 53:151–165
Karrenberg S, Edwards PJ, Kollmann J (2002) The life history of Salicaceae living in the active zone of floodplains. Freshw Biol 47:733–748
Khan MA, Korban SS (2012) Association mapping in forest trees and fruit crops. J Exp Bot 63:4045–4060
Kuzovkina Y, Weih M, Abalos Romero M, Belyaeva I, Charles J, Hurst S, Karp A, Labrecque M, McIvor I, Singh NB et al (2008) Salix: botany and global horticulture. Hortic Revs 34:447–489
Larsson S (1998) Genetic improvement of willow for short-rotation coppice. Biomass Bioenergy 15:23–26
Larsson SJ, Lipka AE, Buckler ES (2013) Lessons from Dwarf8 on the strengths and weaknesses of structured association mapping. PLoS Genet 9(2):e1003246. doi:10.1371/journal.pgen.1003246
Lascoux M, Thorsen J, Gullberg U (1996) Population structure of a riparian willow species, Salix viminalis L. Genet Res 68:45–54
Leinonen T, McCairns RJS, O'Hara RB, Merilä J (2013) Q ST - F ST comparisons: evolutionary and ecological insights from genomic heterogeneity. Nat Rev Genet 14:179–190
Lin J, Gibbs JP, Smart LB (2009) Population genetic structure of native versus naturalized sympatric shrub willows (Salix; Salicaceae). Am J Bot 96:771–785
Marcysiak K (2012) Diversity of Salix reticulata L. (Salicaceae) leaf traits in Europe and its relation to geographical position. Plant Biosyst 146:101–111
Mikola J (1982) Bud-set as an indicator of climatic adaptation of Scots pine in Finland. Silva Fenn 16:178–184
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Pospísková M, Bartáková I (2004) Genetic diversity of a black poplar population in the Morava river basin assessed by microsatellite analysis. For Genet 11:257–262
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rönnberg-Wästljung AC, Samils B, Tsarouhas V, Gullberg U (2008) Resistance to Melampsora larici-epitea leaf rust in Salix: analyses of quantitative trait loci. J Appl Genet 49:321–331
Rousset F (2000) Genetic differentiation between individuals. J Evol Biol 13:58–62
Samils B, Rönnberg-Wästljung AC, Stenlid J (2011) QTL mapping of resistance to leaf rust in Salix. Tree Genet Genomes 7:1219–1235
Savage JA, Cavender-Bares J (2013) Phenological cues drive an apparent trade-off between freezing tolerance and growth in the family Salicaceae. Ecology 94:1708–1717
Savolainen O, Pyhäjärvi T, Knürr T (2007) Gene flow and local adaptation in trees. Annu Rev Ecol Evol Syst 38:595–619
Semerikov V, Lagercrantz U, Tsarouhas V, Rönnberg-Wästljung A, Alström-Rapaport C, Lascoux M (2003) Genetic mapping of sex-linked markers in Salix viminalis L. Heredity 91:293–299
Shafroth PB, Scott ML, Friedman JM, Laven RD (1994) Establishment, sex structure and breeding system of an exotic riparian willow, Salix x rubens. Am Midl Nat 132:159–172
Smulders MJM, Cottrell JE, Lefevre F, van der Schoot J, Arens P, Vosman B, Tabbener HE, Grassi F, Fossati T, Castiglione S et al (2008) Structure of the genetic diversity in black poplar (Populus nigra L.) populations across European river systems: consequences for conservation and restoration. For Ecol Manag 255:1388–1399
Stott KG (1984) Improving the biomass potential of willow by selection and breeding. In: Perttu K (ed) Ecology and management of forest biomass production systems. Swedish University of Agricultural Sciences, Sweden
Trybush SO, Jahodova S, Cizkova L, Karp A, Hanley SJ (2012) High levels of genetic diversity in Salix viminalis of the Czech Republic as revealed by microsatellite markers. Bioenerg Res 5:969–977
Tsarouhas V, Gullberg U, Lagercrantz U (2002) An AFLP and RFLP linkage map and quantitative trait locus (QTL) analysis of growth traits in Salix. Theor Appl Genet 105:277–288
Tsarouhas V, Gullberg U, Lagercrantz U (2003) Mapping of quantitative trait loci controlling timing of bud flush in Salix. Hereditas 138:172–178
Tsarouhas V, Gullberg U, Lagercrantz U (2004) Mapping of quantitative trait loci (QTLs) affecting autumn freezing resistance and phenology in Salix. Theor Appl Genet 108:1335–1342
Viherä-Aarnio A, Häkkinen R, Partanen J, Luomajoki A, Koski V (2005) Effects of seed origin and sowing time on timing of height growth cessation of Betula pendula seedlings. Tree Physiol 25:101–108
Weih M (2009) Genetic and environmental variation in spring and autumn phenology of biomass willows (Salix spp.): effects on shoot growth and nitrogen economy. Tree Physiol 29:1479–1490
Winfield MO, Arnold GM, Cooper F, Le Ray M, White J, Karp A, Edwards KJ (1998) A study of genetic diversity in Populus nigra subsp. betulifolia in the Upper Severn area of the UK using AFLP markers. Mol Ecol 7:3–10
Acknowledgments
The authors acknowledge funding support by the ERANET-Bioenergy scheme grants awarded by the Bioscience and Biotechnology Sciences Research Council (BBSRC) of the UK (BB/G00580X/1) and the Swedish Energy Agency (31468–1).
Data archiving statement
Primer sequences of the SSR markers used in this study are provided in Supplementary Table 2.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Z. Kaya
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
(DOCX 97 kb)
Supplementary Table 2
(DOCX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Berlin, S., Trybush, S.O., Fogelqvist, J. et al. Genetic diversity, population structure and phenotypic variation in European Salix viminalis L. (Salicaceae). Tree Genetics & Genomes 10, 1595–1610 (2014). https://doi.org/10.1007/s11295-014-0782-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-014-0782-5