
Abstract
During the last years it has been an increasing focus on fundamental studies of the Fischer–Tropsch synthesis (FTS). Steady-state isotopic transient kinetic analysis and first principles investigation have proven to be important methods in studying the reaction mechanism of FTS. The present contribution deals with recent progresses in understanding of the F–T mechanism on Co catalysts based on combined DFT calculations, in situ characterization, steady-state isotopic transient kinetic analysis and microkinetics. A brief outlook into future perspectives of the FTS for converting synthesis gas from different carbon sources to fuels and chemicals is also provided.
Graphical Abstract

Similar content being viewed by others

1 Introduction
Fischer–Tropsch synthesis (FTS) is an alternative process for producing transportation fuels and chemicals by converting syngas derived from natural gas, biomass and coal [1–4]. Recently, the increase in demand for transportation fuels and the large reserves of natural gas have made FTS an attractive industrial process. In the FTS process, cobalt-based catalysts, owing to low water–gas shift activity and remarkable stability, is considered to be the favorable catalyst for the synthesis of long-chain hydrocarbons from synthesis gas with a H2/CO ratio about 2 [5–7].
Despite the fact that the FTS process has been in commercial use for decades, several fundamental issues, such as the reaction mechanism, are still under debate. In general, three main mechanisms as displayed in Fig. 1 have gained widespread acceptance [8, 9]. The original carbide mechanism, proposed by Fischer and Tropsch [10, 11], suggested that CO adsorbs dissociatively on the surface and that the surface carbon subsequently is hydrogenated to form methylene (CH2) groups, which is considered as the monomer for the chain propagation. Despite the amount of experimental work that supports this mechanism [12–14], it fails to explain the formation of oxygenates. In respond to the discredit of the carbide mechanism, Anderson and Emmett proposed the hydroxycarbene (enol) mechanism [15, 16], which involves the formation of a hydroxylcarbene intermediate, M-CHOH, responsible for carbon–carbon bond formation. In order to account for the range of products in more detail, Pichler and Schulz developed the CO insertion mechanism [17], in which the insertion of CO into alkyl intermediates represents the chain growth. Unfortunately, none of them can accounts for the full product distribution. Besides, it is possible that the reaction mechanism may change with different reaction conditions and catalysts. There are several excellent reviews regarding the FTS mechanism [1, 2, 18–21]. The reaction mechanism for such a complex reaction is still a project of controversy and uncertainty despite the fact that it has been explored by many experimental and theoretical studies.

Three main mechanisms of Fischer–Tropsch synthesis [22]. a Carbide mechanism. b hydroxycarbene mechanism. c CO insertion mechanism
Transient kinetic studies are particularly helpful in the evaluation of intrinsic kinetic coefficients and understanding reaction mechanisms. Temporal analysis of products (TAP) and the steady-state isotopic transient kinetic analysis (SSITKA) have been applied in kinetic investigations of the F–T synthesis. The approaches of kinetic analysis for FTS will be discussed in Sect. 2. With the advancement of sophisticated computational chemistry techniques, the intricate electronic-structure calculations based on density functional theory (DFT) have recently been widely used to provide fundamental data regarding the surface reaction, which can be a complement to the experimental analysis for understanding of reaction mechanisms [23–26]. More importantly, it can distinguish between those possibilities that were left open, such as various reaction pathways. Since the mechanism of FTS is inconclusive and controversial, more and more DFT calculations have been devoted to tackle these issues [18, 19], especially for the mechanism of CO activation [27–31], methane formation [32–35], and chain growth [36–38]. Despite the successful implementations of DFT in catalysis process, it still holds some drawbacks such as failure in the calculation of weak interactions (such as Van der Waals interaction) [25]. Fortunately, DFT calculations in combination with experimental work (such as spectroscopic methods and kinetic analysis), has proved to be a more powerful tool in modelling active sites and exploring the mechanism at a molecular level. The combined approach has successfully been applied for discriminating the mechanisms of CO activation and methane formation [32, 39–41].
The present review deals with recent progress in understanding the reaction mechanism of FTS on Co catalysts by means of transient kinetic analysis and DFT calculations. The methods of transient kinetic study applied to FTS are summarized, followed by a detailed summary of recent progresses on combined DFT and kinetic analysis of FTS mechanism. The investigation of the FT mechanism has been divided into 4 subtopics: The adsorption of the surface species, CO activation, chain growth, and methane as well as olefin selectivity. In each subtopic, DFT calculations and transient kinetic experiments, which were used to investigate each subtopic, will be reviewed and discussed. The following sections concentrate on the progress from a combined approach of DFT, kinetic analysis and isotope effect. Finally, some conclusions are summarized and a brief outlook into future perspectives is proposed.
2 Approaches of Kinetic Analysis for FTS
The history, applications and recent developments of the temporal analysis of products (TAP) have been reviewed recently [42, 43]. The TAP technique has been applied to different reaction systems involving heterogeneous catalysis: Reaction transport (adsorption and diffusion) [44, 45]; reforming of hydrocarbons [46–51]; hydrogenation and dehydrogenation reactions [52, 53]; environmental catalysis [54–58]; oxidation reactions etc. [59–67]. The TAP technique is useful for obtaining qualitative information about reaction mechanism, such as reaction intermediates and reaction sequences for multistep reactions. It is also possible to obtain quantitative kinetic information by solving the partial differential equations describing chemical and transport processes. There have been limited studies on investigation of FTS using the TAP technique possibly due to the very complicated product spectrum for FTS [68].
Another type of transient experiments is step-response experiments which are much easier to perform and the interpretation of the results is much simpler. This type of experiments involves a step change in reactants, concentration, temperature or pressure. The transient curve of reactants and products are monitored before and after and kinetic information can be extracted from the step-transient signals. Modeling of the response curve is less complicated although the time resolution and sensitivity is lower than for the TAP experiment. The pulse transient method has been used to study the kinetics of several key steps such as CO activation and hydrocarbon chain growth of FTS over supported cobalt catalysts [69, 70].
Steady-state isotopic transient kinetic analysis (SSITKA) combines both isotopic labeling and kinetic analysis. It has contributed significantly to our understanding of a wide range of surface-catalyzed reactions. The review by Shannon and Goodwin has summarized the chemical reactions studied by SSITKA prior to 1995 [71]. Issues explored using SSITKA have included among others the study of mechanisms, the presence and implications of site heterogeneity, the role of chemical promoters and the effect of operation conditions on the surface coverage of intermediates. For FTS, the use of SSITKA has resulted in an improved understanding on the effect of catalyst properties (promoters [72–83], support [84–90], particle size etc. [91–94]) and the reaction conditions [95–101] on the performance. Concerning the mechanism, SSITKA gives the coverage of CO and surface carbon intermediates and the site coverage of H can be estimated. In addition, the site reactivity towards methane and higher hydrocarbons can be obtained. SSITKA can be used to estimate rate coefficients for chain initiation, propagation and termination by fitting theoretically generated model curves to the observed transient responses [102].
SSITKA measures the amount of surface intermediates, but the technique cannot determine the chemical identity of the intermediates. In-situ IR is a powerful technique for identifying surface species at reaction conditions. However, the surface species detected by IR do not necessarily participate in the reaction, may just be a spectator. A powerful tool for identifying active surface intermediates is SSITKA coupled with diffuse reflectance Fourier Transform Infrared Spectroscopy (DRIFS) [103–105]. This coupled technique has been used to investigate formate species in the water–gas-shift reactions [106–108]. However, fewer studies have been carried out to study the mechanism of CO hydrogenation [109].
3 Progresses on Understanding the FTS Reaction Mechanisms by Means of Theoretical and Transit Kinetic Investigations
3.1 Adsorption Energies of the Surface Species
The nature of the adsorbed surface species on cobalt catalysts is crucial for a better understanding of the reaction mechanism, where the configuration and strength of the adsorption directly influence their bond cleavage and formation reactions. Large efforts have been devoted to the studies of adsorption of intermediates on hcp Co (0001), which is the thermodynamically most stable surface of the industrial Co catalysts at FTS condition [110–113]. The chemisorption energies of the most stable configurations for the related surface species on Co (0001) are summarized in Table 1.
Generalized gradient approximation (GGA) was widely used in all studies mentioned in Table 1, which performed better for magnetic surfaces, provided better geometry optimizations and yielded more exact adsorption energies [122, 123]. The chemisorption energies of the same species is slightly different from various sources since different functional (i.e. PBE functional [124] or PW functional [123]), and other parameters were employed in these calculations. Despite this fact, similar trends can be observed as listed in Table 1. For CO adsorption, PW91 [123] and PBE [124] functional provide significantly higher CO chemisorption energies (i.e. 1.76 ± 0.12 eV) on Co (0001) compared to experimental results (i.e. 1.20 ± 0.13 eV) [125, 126]. Moreover, these functional overestimated the stability of highly-coordinated adsorption sites and lead to wrong predictions of the favorable adsorption sites (i.e. the hollow site) [29, 118], while it is reported that CO occupy top sites and form an ordered (\(\sqrt 3 \times \sqrt 3\))R30° structure by LEED pattern [127, 128]. Fortunately, these issues have been tackled with by some correction methods such as the site-dependent correction factors [125, 129], which can provide an adsorption energy in the range of 1.10–1.40 eV and the current methods will be used in future, especially for high CO coverage. It is clearly seen that the chemisorption energies of most C1 species are around or more than 1 eV except for CH3OH, which binds weakly through the O atom since there is no unsaturated atoms in CH3OH [120]. Interestingly, the adsorption energies of n-alkyl with chain length n from 2 to 6 are almost the same because the bonding of these species with the surface is similar. Those of 1-alkenes (n = 2–6) undergo the same trend except for ethylene because the repulsive interaction between the alkyl groups in 1-alkene (n = 3–6) and the surface is stronger than that between H in ethylene and the surface [121]. Noteworthy is that the van der Waals interaction between the 1-alkene and the metal surface, an important contribution to the adsorption energy, is not considered in the exchange–correlation function at PBE level. It has been demonstrated that the chemisorption energy of 1-alkene may play an important role in determining the olefin selectivity and thus more accurate description of the van der Waals interaction is highly desirable [121].
The effects of surface structure and coverage on the adsorption have also been investigated. The adsorption energies on the stepped Co (0001) surface, as shown in Table 1, are larger compared to those on the flat Co (0001) surface. It is demonstrated that defect sites on the step Co (0001) facet lead to lower metal coordination numbers, and as a result the adsorbates binding to the surface are stronger [118, 120]. High binding energies may largely decrease the activation barrier of the bond-breaking steps, indicating that the defect sites on the step surface might be active for CO activation, which will be discussed later. The structure sensitivity of the adsorption process has been further investigated in the refs [113, 115, 118]. It can be seen that the average binding energy of CO is slightly larger for HCP Co (i.e. −1.74 ± 0.088 eV) than that for FCC Co (i.e. −1.66 ± 0.058 eV) [113]. However, the small difference on CO and H adsorption energies between HCP and FCC Co, 0.08 and 0.07 eV, respectively [113], reveals the weak structure sensitivity for the adsorption of the reactant. The effect of CO coverage on the chemisorption of some surface species, H, CO, CH, CH2, CH3, HCO, HCOH, CH2O and so on, has been investigated on Co (0001) with different CO coverages, 1/4, 1/3 and 1/2 ML, as listed in Table 1 [32, 40, 117]. In general, the pre-covered CO molecule decreases the stability of other molecules, especially for larger molecules such as HCOH, due to the repulsive interaction between the adsorbed species. For the CO coverage increasing to 1/2 ML from zero, the chemisorption energy of CH decreased by 0.83 eV, while that of HCOH decreased by 1.08 eV [40]. It should be noted that the hydrogen bond may be formed between the O atom in the pre-covered CO and the O–H in surface species, such as COH, and the stabilizing effect of the hydrogen bond may lead to the slightly enhanced chemisorption by 0.36 eV [32]. In conclusion, the adsorptions of surface species are weakly structure sensitive, while the adsorption energies are affected by the CO coverage (Table 2).
It is worth mentioning that the chemisorption energy is a vital and useful parameter to determine the catalytic activity. As discussed above, the chemisorption energy of 1-alkene may be a controlling parameter in determining the olefin selectivity and thus it is suggested that weakening of the olefin-surface bond will enhance the olefin selectivity [121]. Besides, the linear relationship between the effective barrier of methane formation and the chemisorption of C + 4H(△H) was elucidated by Cheng et al. [35], and a volcano curve to correlate the activity and selectivity to the binding energies of the key intermediate was established, as illustrated in Fig. 2. It is indicated that a good catalyst is a best compromise between activity and selectivity, which require binding energies being on the left side of the volcano curve and close to its top. These correlations may simplify the process of improving the activity and designing better catalyst, which could be of great interest for FTS.

Schematic illustration on the correlation of activity and CH4 selectivity to the binding strength of C + 4H [35]
3.2 CO Activation
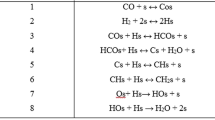
CO activation is the first reaction step in FTS and is critical for the catalytic activity. The issue has been puzzling the scientists ever since the discovery of the FTS, i.e. whether adsorbed CO is directly dissociated to C and O or is hydrogenated to form HCO (or COH) and other intermediates before C=O bond cleavage. Discriminating the two mechanisms is an important issue in FTS. Therefore, a large number of computational catalysis has contributed to the understanding of CO activation through calculations of the activation barriers of the elementary steps involved in different mechanisms, as shown in Table 3.
Hu and coworkers reported that the direct dissociation of CO cannot occur on the flat Co (0001) due to the high barrier of 2.70 eV, while it is much more favorable on the stepped Co (0001) with a barrier of 1.61 eV [29]. Moreover, the barrier of the CO dissociation on the double stepped Co (0001) is even lower of 1.20 eV [130], indicating that the barrier is lower when the surface consists of more defects such as steps and kinks. There are two used methods to construct the DFT models containing defects, where one is creating the defect by removing certain atoms on the low Miller index orientations (such as stepped Co (0001)) [33, 36, 116, 120] and the other is using the model based on the high Miller index orientations that produce corrugated surfaces [113, 118, 131]. Ge et al. [118] studied the effect of defects on CO dissociation by employing the Co (11–20), Co (10–12) and Co (11–24) as model surfaces shown in Fig. 3, in which Co (11–20) is more open and corrugated than Co (0001), Co (10–12) contains steps and Co (11–24) contains double steps and kinks. As expected, the barrier of CO dissociation decreases in the order Co (11–20) > Co (10–12) > Co (11–24) as shown in Table 3, owing to the fact that the defect sites provide stronger binding sites for C and O adatoms and facilitate the CO dissociation. As displayed in Table 3, the activation energies of direct dissociation (i.e. CO → C + O) on the flat Co (0001) which are in the range of 2.28–3.80 eV are dramatically higher than those on surfaces with defects which are in the range of 0.70–2.02 eV, indicating that CO direct dissociation is difficult on ideal Co (0001) and highly sensitive to the local structure. It can be deduced that special active sites on stepped or corrugated surfaces account for the CO direct dissociation. Along this line, van Santen and coworkers proposed that the active sites, so-called B5, is necessary for direct CO dissociation by minimizing the bond competition [1]. B5 sites contain four metal atoms in one layer and a fifth atom in lower layer and usually exist on stepped surfaces. Recently, a novel active site named six-fold (F6) site for one-step CO dissociation was reported on Co (10–10) surface by the same group [28], with four metal atoms at the same layer and two below the layer. The barrier of CO dissociation is 0.70 eV on F6, which is the lowest, reported till now among these surfaces. Thus, CO direct dissociation is highly structure sensitive and the active sites (i.e. B5 and F6) are essential for CO direct dissociation, which indicates that it is very difficult to take place on the ideal Co (0001) surface.

The top view and the side view of the surfaces Co (11–20) (left), Co (10–12) (middle) and Co (11–24) (right). The first layer is  , the second layer
, the second layer  , the third layer
, the third layer  and the forth layer
and the forth layer 
Although direct dissociation of CO is kinetically challenging on flat Co (0001) surface, an alternative pathway via HCO and CH2O was proposed by Holmen and coworkers [132]. The activation energies of all the elementary steps were estimated by using the unity bond index-quadratic exponential potential (UBI-QEP) method and two pathways (one is via COH and the other is via HCO) were established by comparing the activation energies. Furthermore, based on the elementary reaction rates predicted by microkinetic modelling, the favorable mechanism (i.e. the pathway via HCO) was proposed, and HCO* + H* → CH2O* + * was suggested to be the rate determining step. This mechanism has since then been investigated by many using DFT calculations. Inderwildi et al. found the same pathway via HCO and CH2O molecules, which undergo a lower barrier of 0.85 eV for C–O bond cleavage of CH2O [131]. Besides, the similar results of C–O bond cleavage (0.93 eV via HCO and 0.70 eV via CH2O) were obtained by Saeys and coworkers [114]. Moreover, Iglesia and coworkers reported that the C–O bond cleavage barrier via HCO and HCOH is 1.43 and 0.98 eV, respectively on flat Co (0001) with 1/2 ML pre-covered CO [40]. Chen and coworkers also found that C=O bond cleavage undergo barrier of 0.90, 0.73, 0.95 eV via HCO, HCOH and CH2O, respectively on Co (0001) with 1/4 ML pre-covered CO [32]. It is concluded that CO hydrogenation weakens the C–O bond and facilitates the bond scission. Therefore, the hydrogen-assisted CO activation mechanism is the dominating pathway on a flat Co (0001) surface. However, extremely short-lived formyl species are difficult to be spotted under realistic conditions, which calls for a femtosecond spectroscopy apparatus to detect the species and provide the experimental support. Furthermore, Liu et al. [115] elucidated that the barriers of hydrogen-assisted CO activation are 0.73, 1.04 and 0.72 eV on Co (0001), Co (10–12) and Co (11–20) surfaces, respectively indicating that the hydrogen-assisted CO dissociation is also a structure sensitive reaction which depends on the local structure of the active sites.
As discussed above, the hydrogen-assisted CO dissociation is more kinetically favorable on the flat Co (0001) surface compared with the direct dissociation, while the overall barrier of the pathway via HCO (1.27 eV) is slightly lower than that of the direct dissociation (1.47 eV) on Co (211) [133]. For Co (10–12) and Co (11–20), the overall barrier of H-assisted CO dissociation (1.40 and 1.29 eV) is similar to those of direct CO dissociation (1.34 and 1.39 eV) [115]. However, it is demonstrated recently by van Santen and coworkers that the hydrogen-assisted pathway is unfavorable for the Co (10–10) surface since the overall barrier of the hydrogen-assisted pathway via HCO is 0.38 eV higher than the direct dissociation and COH is thermodynamically unstable [28]. Based on this, they proposed that the carbide mechanism is more favorable [27]. Recently, Li and coworkers employed first-principles kinetic studies to identify the structure-sensitivity of the CO dissociation in FTS [113]. Firstly, based on the Wulff construction, Co (11–21), Co (10–11), Co (10–12) and Co (11–20) were suggested to represent hcp Co, while Co (100), Co (311) and Co (110) represent fcc Co. It has been reported that hcp Co is more active than fcc Co, and that the direct dissociation is prevailing for hcp Co while the hydrogen-assisted pathway is prevailing for fcc Co. This may provide new insight for designing better catalyst; it is, however, important to note that the metal-reactant/intermediate interaction and metal-support interaction may lead to rearrangement of the surface, which means that the dominating surfaces for hcp Co and fcc Co may change and correspondingly effect the mechanism. Moreover, whether the hcp or fcc phase is dominating in FTS depends on the activation conditions [134, 135] and the rearrangement of the surface during real FT reaction condition has been demonstrated by both experimental and theoretical methods [136, 137]. It is therefore hard to verify which phase (i.e. hcp or fcc) plays the most important mole as catalyst during the reaction.
It is widely accepted that the CO activation is structure sensitive, leading to the different preferred mechanism on various surfaces. Meanwhile, the rearrangement of the cobalt-based catalyst can change the local structure and may create new active sites, resulting in more complicated mechanisms. Despite the different proposals regarding the CO activation mechanism, it is agreed that hydrogen-assisted CO activation is more kinetically favorable on ideal Co (0001). It is reported that hcp Co catalysts show higher FTS activity than fcc Co [134, 135, 138–141]. Moreover, the larger sized Co catalysts, exposing mainly Co (0001) facets, exhibit higher FTS intrinsic reactivity compared to the smaller sized Co catalysts below 6 nm, possibly because the latter has strongly bonded carbon and oxygen surface species acting as site blocking species, and small particles may also be easily oxidized by the water vapor [6, 92, 142], suggesting a large contribution of Co (0001) to the reaction under reaction condition. Step-transient (chemical transient) experiments have also been used to study CO activation for FTS [69]. A switch form Ar or Ar/H2 to CO/He at reaction temperature (493 K) for a reduced cobalt catalyst showed a delay of the appearance of the CO transient curve in the case of (Ar/H2 → CO/He), suggesting that the presence of H2 increases the amount of CO adsorption and dissociation. The back switch (CO/He → Ar/H2) at 493 K produced methane prior to the formation of CO2. This provides experimental evidence for the conclusion that the dissociation of CO in the presence of H2 proceed through concerted mechanism. By considering the DFT calculations and experimental evidences as discussed above, the hydrogen-assisted CO dissociation is shown to be more kinetically favorable on cobalt-based catalyst. However, it should be noted that a high CO coverage (about 0.5 ML) has been found in steady state isotopic transient kinetic analysis (SSITKA) studies at FT conditions [6], indicating that further studies on the surface with high CO coverage are highly encouraged.
3.3 Chain Growth
The FTS is a complicated reaction system consisting of a large number of intermediates and products, such as paraffins, olefins, oxygenates. Understanding the mechanism of chain propagation and termination is crucial for the production of desired products.
Although it is difficult to simulate the elementary steps involving large molecules on the catalyst surface due to the limitation of current computational capacity, some theoretical efforts [36, 37, 116, 119, 120, 143] have been devoted to this field concerning the formation of C2 and C3. Hu and coworker have investigated all the possible reaction pathways for C1 + C1 coupling for the carbide mechanism based on DFT calculations and kinetic analyses [116]. As shown in Table 4, the barriers for all C1 + C1 coupling pathways on the flat surface are smaller than those on the stepped surface except for CH2 + CH2 and CH2 + CH3. However, taking into account the differences in adsorption energies of the adsorbed reactant on the flat and stepped surface in the calculations of reaction rates, it is identified that the C + C coupling reaction proceeds mainly on the step sites and that CH3 + C and CH2 + CH2 are two major chain growth pathways at the step sites based on the calculated reaction rates for all C1 + C1 coupling pathways. Hu and coworkers [36] then studied the behavior of C2 hydrogenation, C2 + C1 and C3 + C1 coupling reactions in detail on stepped Co (0001) to understand the trend of the chain growth and termination with different chain length. C2 + C1 and C3 + C1 coupling reactions give similar geometries of the TSs (Transition States) as the corresponding C1 + C1 coupling reactions. A minor variation from C1 + C1 is that the barrier of the reactions of CH3C + C and CH3C + CH are 0.38 and 0.32 eV smaller than the corresponding C1 + C1 coupling, indicating that the other two RC + C and RC +CH may also contribute to the chain growth. Except for those two steps, the activation barriers of other C2 + C1 are similar to the corresponding C1 + C1 coupling, and the coupling reactions of RCH2 + C and RCH + CH2 are still the most possible chain growth pathways for C2 + C1 coupling based on theoretical kinetic analysis. Furthermore, it is verified that the variations of the barriers among the C–C coupling reactions are quite small when the chain length n > 2 by comparing the barriers of the CH3CH2C + C and CH3CH2C + CH (1.63 and 1.47 eV, respectively) with those of CH3C + C and CH3C + CH (1.58 and 1.44 eV, respectively) and it is expected that the chain growth mechanism is invariable with different chain lengths. Hence, RCH2 + C and RCH + CH2 may be two major pathways for all the C + C coupling with chain lengths n≧2 in the carbide mechanism, which is consistent with the experimental results of Brady and Pettit as discussed below [12, 13]. In the 1980s, Robert C. Brady III and R. Pettit used diazomethane, a molecule believed to provide CH2 group on the catalyst surface to distinguish between the three major mechanisms. The CH2 plays different roles in the three mechanisms. CH2 participates both in chain initiation and chain growth in the carbide mechanism, CH2 is only initiating in the CO insertion mechanism; and CH2 are not involved in the hydroxyl methylene mechanism. Using 12CH2N2, 13CO and H2, the isotopic content was monitored for propane formation. The carbide mechanism should produce all kinds of combination with 12C and 13C in propane; the CO insertion mechanism should produce 12C–13C–13C and 13C–13C–13C and the hydroxyl mechanism should produce 13C–13C–13C. Based on the composition of the produced propane, it was concluded that only the carbide mechanism could produce the product compositions and the other mechanisms were therefore excluded. Although monomer is generated through dissociation of CO the exact composition is not known, whether it is C, CH or CH2 specie. However, it should be pointed out that an assumption was automatically made in their analysis, where the reaction of CH2 with O and OH were ignored. If the reaction steps of 11 and 13 in Table 3 are in equilibrium, the conclusion could be not conclusive.
Recently, Schouten and co-workers have combined experimental data and kinetic modeling to further identify and discriminate between different mechanistic models on cobalt based catalysts for the FTS [145–147]. The prerequisite is that different mechanism models have to be proposed first before model discrimination by comparing with experimental data. It was concluded that the formation of hydrocarbons proceeds via a two-pool mechanism, where two carbon pools contribute to methane formation and C–C coupling. The most abundant chemisorbed surface species are COads and two single-C species, Cα;ads and Cβ;ads. Besides, the fractional surface coverage of growing hydrocarbon chains is as low as 1.4 %. The rate coefficient for chain initiation is one order of magnitude lower than that for chain growth, indicating that CO activation is the rate determining step in the overall CO conversion. It is also observed that the rate coefficient for ethene readsorption is one order of magnitude higher than for readsorption of higher 1-olefins, which can explain the lower ethylene/ethane selectivity. Chain branching and bond shift are important secondary reactions at atmospheric pressure, transforming reactive 1-olefins into unreactive internal-olefins and iso-olefins, thus decreasing the asymptotic chain growth probability. The termination to paraffins is expected to be represented by a hydrogenation reaction. The termination to olefin, however, appears to be a desorption step rather than a hydrogenation or a dehydrogenation reaction. The growing hydrocarbon chain is therefore represented by a CiH2i species.
As an alternative to the carbide mechanism, the CO insertion mechanism on cobalt-based catalysts has been investigated by Saeys and coworkers [114]. As shown in Table 5, it is reported that the barriers of CO insertion into CH2 and CH are 1.11 and 0.83 eV respectively, which is comparable to those of the C1 + C1 coupling. However, the calculated TOF is still lower than the experimental TOF, owing to the effect of CO coverage on the energetic data that is not taken into account. Furthermore, the effect of CO coverage on the CO insertion mechanism was studied [117]. The higher CO coverage decreased the activation barrier for the CH2 + CO coupling by 0.24 eV, while it increased the barriers for the C–O scission reactions. However, it is necessary to consider the complete pathways rather than the activation barrier of individual steps. By analyzing the energy profiles of the complete pathways, a favorable pathway were proposed, namely that the RC (R represent hydrogen or an alkyl) group is hydrogenated to RCH, which then undergo CO insertion and hydrogenation to RCH2CO [117]. The calculated CO TOF of this mechanism is quite close to the experimental value, indicating that the calculations on Co (0001) surface with high CO coverage are more accurate and that the CO insertion mechanism could be facile on cobalt-based catalysts.
The experimental evidence for the CO insertion mechanism has derived from chemical transient kinetic studies, which has been used to provide surface coverage of C, O, H species for cobalt based FTS [148, 149]. This type of experiment is performed by switching form one reactant/inert to both reactants and after reaching steady-state switching back to one reactant/inert. The relaxation of the reactants and products during the whole process was monitored by MS.
Figure 4 shows the build-up transient, in which methane production runs through a maximum, and CO appears at that very moment. Moreover, C2+ hydrocarbon formation does not start before both the occurrence of CO and the maximum in CH4 production. This observation provides a first indication for CO involved in C2+ formation. However, further DFT and experimental studies are needed to distinguish the nature of the monomer such as CO and CHxOy, in the chain growth. Moreover, the C2+ hydrocarbons appear in sequence; i.e., C2H6 is formed first, followed shortly by C3H8 and C4H10, confirming the polymerization reaction mechanism where a C1 monomer is added at a time.

The flux-out at the outlet of the reactor during the build-up stage on a Co/MgO catalyst at 503 K (H2/inert → H2/CO) [70]
Figure 5 also shows that, once the CO inlet flow stops, the C2+ hydrocarbon (and CO2) production decreases very quickly (after passing a short-term maximum of ∼1.5 times their steady-state values). According to the authors, this also provides a strong indication for weakly adsorbing CO playing the role of the C1 monomer. Based on the mass balance, the site coverage of surface C, O species could be followed at any reaction time. The authors found that the chain growth probability is not proportional to the surface C coverage but to the partial pressure of CO, a result that further supports their argument that CO is the chain lengthening monomer. This example shows that transient experiments could provide useful information regarding reaction mechanisms, although the results need a carful interpretation.

The issue regarding the chain growth mechanism, whether the carbide mechanism or CO insertion mechanism is the more favorable, has not yet been solved since both mechanisms have theoretical and experimental support. More systematic studies are required to elucidate the monomer in the chain growth in FTS.
3.4 Methane and Olefin Selectivity
3.4.1 Methane Selectivity
A major disadvantage of Co-based FT catalysis is the relatively high selectivity of methane, deviating from the ideal ASF distribution. If syngas is derived from natural gas, methane is the least desired byproduct in the FTS, and understanding the mechanism for CH4 formation is therefore important to suppress its selectivity. A few theoretical efforts have been devoted to the mechanism of CH4 formation and the activation energies for CHx hydrogenation on different surfaces are summarized in Table 6.
As shown in Table 6, Hu and coworkers reported that CH3 hydrogenation was the kinetically controlling step with barrier around 1 eV in CHx(x = 0–3) hydrogenation on both flat and stepped Co (0001) surfaces, while the other three steps have barriers below 0.90 eV [33]. CH2 is found to be the most unstable species among the CHx(x = 1–3) species since it could decompose to CH or be hydrogenated to CH3 with low barriers on both surfaces. Furthermore, they extended the studies to Rh, Ru, Fe and Re catalysts [35] and suggested that the effective barrier difference between methane formation and chain growth (△Eeff) is an energy descriptor for quantifying the selectivity. In addition, a linear relationship between △Eeff and the binding energy of C + 4H was derived, which has been illustrated in Sect. 3.1. The CHx(x = 0–3) hydrogenation on Co2C (001) as also studied by Cheng et al. [144] and they reported that the methane selectivity is higher on cobalt carbides compared with metallic Co, indicating that metallic cobalt is the preferred active site for FTS. Recently, similar geometrical and energetic information of CHx(x = 0–3) hydrogenation were also derived for Co (0001), Co (10–12) and Co (11–20) by Li and coworkers [115], indicating the CHx(x = 0–3) hydrogenation are not significantly affected by the local structure. It should be noted that CHX is generated via CO activation and thus the structural sensitivity for the methanation reaction is ascribed to the structure sensitivity of CO activation. Recently, a systematic investigation of all possible reaction routes for methane formation starting from the gas phase CO is studied by a combined approach of DFT calculations and kinetic studies [32], and the favorable route is suggested to be that CO undergoes hydrogenation to form HCOH and then HCOH is dissociated to CH, which will be hydrogenated further to form CH4. The detailed discussion will be carried out in Sect. 4.
3.4.2 Olefin Selectivity
Lower olefins, such as ethylene, propylene and butylene, are key building blocks in the chemical industry. FTS could be an alternative production route for these high-value commodity chemicals and investigating the olefin selectivity is therefore of great interest. The hydrogenation and dehydrogenation of n-alkyl groups on Co (0001) were studied, and the corresponding barriers are listed in Table 7 [121]. It can be found that the transition states and the activation barrier for CnH2n (n ≥ 2) hydrogenation are almost the same, and that those for CnH2n+1 (n ≥ 2) hydrogenation are also the same. The higher ethane/ethylene ratio is attributed to the 0.2 eV larger adsorption energy of ethylene, which is consistent with experimental results showing that the rate coefficient of ethene readsorption is one order of magnitude higher than for readsorption of higher 1-olefins [145]. Besides, the chain length dependence of the paraffin/olefin ratio is determined by both the van der Waals interaction between α-olefin and the metal surface and the entropy difference between adsorbed and gaseous α-olefin [121]. Thus, in order to get more accurate energetic information, the higher level functional including van der Waals interaction is important for these calculations.
4 A Combined Approach of DFT Calculations and Kinetic Analysis
No single technique can exclusively elucidate the reaction mechanism for such A complex reaction. Recently, a combined approach of theoretical calculations and experimental kinetics has been employed to discriminates between the different mechanisms of CO activation and methane formation on cobalt-based catalysts [32, 39, 41]. In 2010, Iglesia and coworkers observed the inverse isotope effect on Co catalysts, which is due to the compensation between the thermodynamic (H2 dissociation to H*, H* addition to CO* species to form HCO*) and kinetic (H* reaction with HCO*) isotope effect. It is also shown that the calculated kinetic isotope effect of the hydrogen-assisted CO dissociation pathway is quite close to the experimentally one, indicating that the H-assisted CO activation prevails on Co catalyst [41].
Yang et al. employed combined DFT, transient, and steady-state kinetic modeling to elucidate the CO activation mechanism [39]. The combined approach of DFT calculations and kinetic analysis are illustrated in Fig. 6. On one hand, SSITKA studies can provide information about the coverage and surface residence time of surface intermediates and other key parameters, which are useful for kinetic studies. More importantly, the kinetic isotope effect can be derived by D2/H2 isotopic studies. On the other hand, DFT calculations can give energetic information of the reaction, such as activation energy, and it can obtain kinetic and equilibrium isotopic effects of the elementary steps through calculating the vibration frequencies of the ISs (initial states), TSs (transition states) and the FSs (final state). In order to discriminate different possible pathways, the DFT-based kinetic isotope effects (KIE) and kinetic parameters (i.e. the reaction order of reactants and reaction rates), were compared with the corresponding experimental results from SSTKA analyses. This opens a new window to comprehensively discriminate and identify possible reaction pathways. The authors coupled the transient and steady state modeling to derive Langmuir–Hinshelwood type kinetic equations, which can predict very well the experimental TOF for CO conversion by taking into account the effect of CO coverage on the CO equilibrium (KCO). However, it is difficult to exclusively elucidate the reaction mechanisms based solely on kinetic modeling, such as direct CO dissociation with C + H → CH as RDS and hydrogen-assisted CO activation with either HCO + H → HCOH or HCOH → CH + OH as RDS. Together with DFT investigations of the kinetic isotopic effect, hydrogen-assisted CO dissociation is demonstrated to be the dominating CO activation pathway [39]. Two carbon pools, CH2O and CHX, have also been identified for methane formation. By a similar combined approach, the mechanism of methane formation was discriminated among nine possible reaction pathways [32]. The DFT calculations were first performed and the activation energies and free energies were derived. Only three pathways were energetically unfavorable based on the DFT calculations. For the six possible pathways, the reaction orders of the derived Langmuir–Hinshelwood (L–H) rate expressions were compared with experimental results, and the results showed that three pathways could not be discriminated since they all fit well with the experimental data. Furthermore, the isotope effects of the remaining three ones were calculated by DFT and compared with the experimental value. Finally, one pathway was suggested to be most favorable, that is, CO undergoes hydrogenation to form HCOH and then HCOH is dissociated to CH, which will be hydrogenated further to form CH4. It is demonstrated that the different pathways cannot be discriminated solely by one method and that the combined approach of DFT, kinetic analysis and KIE is a powerful tool to identify reaction mechanisms. This is expected to be used for further studies, such as chain growth mechanism, and also be extended to other catalysis systems.

Schematic illustration of the combined approach of DFT calculations and kinetic analysis
5 Conclusions and Perspectives
FTS is a complex reaction involving a large number of surface species and elementary steps for the formation of products. It has been demonstrated that theoretical analysis and transient kinetic studies are powerful tools to provide fundamental insight into FTS mechanism on cobalt catalysts. Some key conclusions of this present review are highlighted and summarized as follows.
It is widely accepted that CO activation is a structure sensitive reaction. For instance, the activation energy of direct CO dissociation is much lower on stepped surfaces including the active sites (such as B5 or F6) than on ideal Co (0001) surface. For the ideal Co (0001) surface, hydrogen-assisted CO activation is the preferred pathway. For the stepped surfaces, there are still debates concerning the preferred CO activation pathway since different surface model, different CO coverage and different evaluation criteria (e.g., the activation barriers of elementary steps, the overall barriers, effective barriers and calculated reaction rates) are used. Considering that the small sized particles with more stepped surfaces are easily blocked by strong bonded carbon and oxygen species, the hydrogen-assisted CO dissociation mechanism seems to be more favorable, which is also consistent with step-transient experimental results.
As for the chain growth mechanism, both the carbide mechanism and the CO insertion mechanism seem to be possible. In the carbide mechanism, RCH2 + C and RCH + CH2 are the two major pathways for C + C coupling with chain length n≧2 based on the DFT calculations. In the CO insertion mechanism, the pathway involving RC group hydrogenation to RCH, then CO insertion and hydrogenation to RCH2CO, was proposed based on the DFT calculations. However, a systematic study including both experimental and theoretical investigations is highly desired to provide a better understanding of chain growth monomer, chain growth probability and olefin to paraffin formation of hydrocarbons with different carbon numbers.
Regarding methane formation, the two carbon pools, CH2O and CHX, were found responsible for methane formation by transit kinetic modelling. The effective barrier difference between methane formation and chain growth (△Eeff), is regarded as an energy descriptor for quantifying the selectivity. Combined approach of kinetic analysis, kinetic isotope effect analysis and DFT calculations shows that the preferred methane formation pathway is CO hydrogenation to form HCOH, then HCOH dissociation to CH and subsequent hydrogenation to form CH4. For α-olefin selectivity, the adsorption energies of olefins (especially the van der Waals interaction between α-olefin and the metal surface and the entropy difference between the absorbed and gaseous α-olefin) determine the chain length dependence of the paraffin/olefin ratio. However, van der Waals interaction was not involved, calling for further studies.
The methodology, combining DFT calculations, kinetic isotopic effect and kinetic analysis, has been successfully used to discriminate between different proposals for the mechanism of CO activation and methane formation. It is expected that using the method will made it possible to further elucidate the chain growth and the olefin selectivity. This method could be applicable for mechanistic studies of other complex catalytic systems. It should be noted that realistic FTS reaction conditions such as temperature, pressure and CO coverage of course need to be concerned, and that a better evaluation criteria, among others activation barriers, free energies, effective barrier and reaction rates, should be developed.
References
van Santen RA, Ciobica IM, van Steen E, Ghouri MM (2011) In: Gates BC, Knozinger H (eds) Advances in catalysis, vol 54. Elsevier, San Diego, p 127
van Steen E, Claeys M (2008) Chem Eng Tech 31:655
Biloen P, Sachtler WMH (1981) Adv Catal 30:165
Huber GW, Iborra S, Corma A (2006) Chem Rev 106:4044
Yang J, Tveten EZ, Chen D, Holmen A (2010) Langmuir 26:16558
den Breejen JP, Radstake PB, Bezemer GL, Bitter JH, Froseth V, Holmen A et al (2009) J Am Chem Soc 131:7197
Bezemer GL, Bitter JH, Kuipers H, Oosterbeek H, Holewijn JE, Xu XD et al (2006) J Am Chem Soc 128:3956
Dry ME (1996) Appl Catal A 138:319
Roferdepoorter CK (1981) Chem Rev 81:447
Fischer F, Tropsch H (1926) Chem Ber 59:830
Fischer F, Tropsch H (1926) Brennstoff-Chem 7:97
Brady RC, Pettit R (1981) J Am Chem Soc 103:1287
Brady RC, Pettit R (1980) J Am Chem Soc 102:6181
van Barneveld WAA, Ponec V (1984) J Catal 88:382
Kummer JT, Emmett PH (1953) J Am Chem Soc 75:5177
Storch HH, Golumbic N, Anderson RB (1951) The Fischer–Tropsch and Related syntheses. Wiley, New York
Pichler H, Schulz H (1970) Chem Ing Tech 42:1162
Cheng J, Hu P, Ellis P, French S, Kelly G, Lok CM (2010) Top Catal 53:326
Valero MC, Raybaud P (2013) Catal Lett 143:1
van Santen RA, Markvoort AJ, Filot IAW, Ghouri MM, Hensen EJM (2013) PCCP 15:17038
van Santen RA, Ghouri MM, Shetty S, Hensen EMH (2011) Catal Sci Technol 1:891
Hindermann JP, Hutchings GJ, Kiennemann A (1993) Catal Rev 35:1
Kohn W, Becke AD, Parr RG (1996) J Phys Chem 100:12974
Orio M, Pantazis DA, Neese F (2009) Photosynth Res 102:443
Sousa SF, Fernandes PA, Ramos MJ (2007) J Phys Chem A 111:10439
Cramer CJ, Truhlar DG (2009) PCCP 11:10757
Shetty S, van Santen RA (2011) Catal Today 171:168
Shetty S, van Santen RA (2010) PCCP 12:6330
Gong XQ, Raval R, Hu P (2004) Surf Sci 562:247
Liu ZP, Hu P (2001) J Chem Phys 114:8244
Pham TH, Duan X, Qian G, Zhou X, Chen D (2014) J Phys Chem C 118:10170
Qi Y, Yang J, Duan X, Zhu Y-A, Chen D, Holmen A (2014) Catal Sci Technol 4:3534
Gong XQ, Raval R, Hu P (2005) J Chem Phys 122:024711
Huo C-F, Li Y-W, Wang J, Jiao H (2009) J Am Chem Soc 131:14713
Cheng J, Hu P, Ellis P, French S, Kelly G, Lok CM (2009) J Phys Chem C 113:8858
Cheng J, Hu P, Ellis P, French S, Kelly G, Lok CM (2008) J Catal 257:221
Cheng J, Hu P, Ellis P, French S, Kelly G, Lok CM (2008) J Phys Chem C 112:6082
Ciobica IM, Kramer GJ, Ge Q, Neurock M, van Santen RA (2002) J Catal 212:136
Yang J, Qi Y, Zhu J, Zhu Y-A, Chen D, Holmen A (2013) J Catal 308:37
Ojeda M, Nabar R, Nilekar AU, Ishikawa A, Mavrikakis M, Iglesia E (2010) J Catal 272:287
Ojeda M, Li AW, Nabar R, Nilekar AU, Mavrikakis M, Iglesia E (2010) J Phys Chem C 114:19761
Gleaves JT, Yablonsky G, Zheng X, Fushimi R, Mills PL (2010) J Mol Catal A 315:108
Pérez-Ramírez J, Kondratenko EV (2007) Catal Today 121:160
Wang D, Dewaele O, Froment GF (1998) J Mol Catal A 136:301
Keipert OP, Baerns M (1998) Chem Eng Sci 53:3623
Schuurman Y, Marquez-Alvarez C, Kroll VCH, Mirodatos C (1998) Catal Today 46:185
Schuurman Y, Mirodatos C, Ferreira-Aparicio P, Rodríguez-Ramos I, Guerrero-Ruiz A (2000) Catal Lett 66:33
O’Connor AM, Schuurman Y, Ross JRH, Mirodatos C (2006) Catal Today 115:191
Duarte RB, Olea M, Iro E, Sasaki T, Itako K, van Bokhoven JA (2014) ChemCatChem 6:2898
Bachiller-Baeza B, Mateos-Pedrero C, Soria MA, Guerrero-Ruiz A, Rodemerck U, Rodriguez-Ramos I (2013) Appl Catal B 129:450
Hevia MAG, Bridier B, Pérez-Ramírez J (2012) Appl Catal A 439–440:163
Bron M, Kondratenko E, Trunschke A, Claus P (2004) Int J Res Phys Chem Chem Phys 218:405
Morgan K, Goguet A, Hardacre C, Kondratenko EV, McManus C, Shekhtman SO (2014) Catal Sci Technol 4:3665
Setiabudi A, Chen J, Mul G, Makkee M, Moulijn JA (2004) Appl Catal B 51:9
Kondratenko EV, Kondratenko VA, Richter M, Fricke R (2006) J Catal 239:23
Kondratenko E, Pérez-Ramírez J (2003) Catal Lett 91:211
Kabin KS, Khanna P, Muncrief RL, Medhekar V, Harold MP (2006) Catal Today 114:72
Kondratenko VA, Baerns M (2004) J Catal 225:37
Pérez-Ramírez J, Kondratenko EV, Kondratenko VA, Baerns M (2004) J Catal 227:90
Schäffer J, Kondratenko VA, Steinfeldt N, Sebek M, Kondratenko EV (2013) J Catal 301:210
Beck B, Fleischer V, Arndt S, Hevia MG, Urakawa A, Hugo P et al (2014) Catal Today 228:212
Martin GA, Mirodatos C (1995) Fuel Process Technol 42:179
Menon U, Galvita VV, Marin GB (2011) J Catal 283:1
Nijhuis TA, Makkee M, van Langeveld AD, Moulijn JA (1997) Appl Catal A 164:237
Daniells ST, Overweg AR, Makkee M, Moulijn JA (2005) J Catal 230:52
Redekop EA, Yablonsky GS, Constales D, Ramachandran PA, Gleaves JT, Marin GB (2014) Chem Eng Sci 110:20
Widmann D, Hocking E, Behm RJ (2014) J Catal 317:272
Hong J, Pietrzyk S, Khodakov AY, Chu W, Olea M, Balcaen V et al (2010) Appl Catal A 375:116
Dai X, Yu C (2008) J Nat Gas Chem 17:365
Schweicher J, Bundhoo A, Kruse N (2012) J Am Chem Soc 134:16135
Shannon SL, Goodwin JG (1995) Chem Rev 95:677
Schanke D, Vada S, Blekkan EA, Hilmen AM, Hoff A, Holmen A (1995) J Catal 156:85
Yang J, Chen D, Holmen A (2012) Catal Today 186:99
Vada S, Hoff A, Adnanes E, Schanke D, Holmen A (1995) Top Catal 2:155
Kogelbauer A, Goodwin JJG, Oukaci R (1996) J Catal 160:125
Panpranot J, Goodwin JG Jr, Sayari A (2002) J Catal 211:530
Bertole CJ, Mims CA, Kiss G (2004) J Catal 221:191
Bertole CJ, Kiss G, Mims CA (2004) J Catal 223:309
Haddad GJ, Chen B, Goodwin JJG (1996) J Catal 161:274
Vada S, Chen B, Goodwin JG (1995) J Catal 153:224
Vada S, Kazi AM, Bedu-Addo FK, Chen B, Goodwin JG Jr (1994) In: Curry-Hyde HE, Howe RF (eds) Studies in surface science and catalysis. Elsevier, Amsterdam, p 443
den Breejen J, Frey A, Yang J, Holmen A, van Schooneveld M, de Groot FF et al (2011) Top Catal 54:768
Borg Ø, Hammer N, Enger BC, Myrstad R, Lindvåg OA, Eri S et al (2011) J Catal 279:163
Rohr F, Lindvåg OA, Holmen A, Blekkan EA (2000) Catal Today 58:247
Jongsomjit B, Panpranot J, Goodwin JG Jr (2003) J Catal 215:66
Enger BC, Frøseth V, Yang J, Rytter E, Holmen A (2013) J Catal 297:187
Enger BC, Fossan Å-L, Borg Ø, Rytter E, Holmen A (2011) J Catal 284:9
Tsai Y-T, Mo X, Goodwin J Jr (2012) Top Catal 55:757
Rane S, Borg Ø, Yang J, Rytter E, Holmen A (2010) Appl Catal A 388:160
Phan X, Yang J, Bakhtiary-Davijnay H, Myrstad R, Venvik H, Holmen A (2011) Catal Lett 141:1739
den Breejen JP, Radstake PB, Bezemer GL, Bitter JH, Frøseth V, Holmen A et al (2009) J Am Chem Soc 131:7197
Yang J, Tveten EZ, Chen D, Holmen A (2010) Langmuir 26:16558
Mo X, Tsai Y-T, Gao J, Mao D, Goodwin JG Jr (2012) J Catal 285:208
Carballo JMG, Yang J, Holmen A, García-Rodríguez S, Rojas S, Ojeda M et al (2011) J Catal 284:102
Frøseth V, Holmen A (2007) Top Catal 45:45
Panpranot J, Goodwin JG Jr, Sayari A (2003) J Catal 213:78
Rothaemel M, Hanssen KF, Blekkan EA, Schanke D, Holmen A (1997) Catal Today 38:79
Hanssen KF, Blekkan EA, Schanke D, Holmen A (1997) In: Froment GF, Waugh KC (eds) Studies in surface science and catalysis. Elsevier, Amsterdam, p 193
Bertole CJ, Mims CA, Kiss G (2002) J Catal 210:84
Frøseth V, Storsæter S, Borg Ø, Blekkan EA, Rønning M, Holmen A (2005) Appl Catal A 289:10
Krishnamoorthy S, Tu M, Ojeda MP, Pinna D, Iglesia E (2002) J Catal 211:422
Krishna KR, Bell AT (1993) J Catal 139:104
Schweicher J, Bundhoo A, Frennet A, Kruse N, Daly H, Meunier FC (2010) J Phys Chem C 114:2248
Meunier FC, Tibiletti D, Goguet A, Burch R (2006) Oil Gas Sci Technol 61:497
Chansai S, Burch R, Hardacre C, Breen J, Meunier F (2010) J Catal 276:49
Jacobs G, Davis BH (2007) Appl Catal A 333:192
Kalamaras CM, Olympiou GG, Efstathiou AM (2008) Catal Today 138:228
Savva PG, Costa CN, Efstathiou AM (2008) Kinet Catal 49:743
Schweicher J, Bundhoo A, Frennet A, Kruse N, Daly H, Meunier FC (2010) J Am Chem Soc 114:2248
Lee BW, Alsenz R, Ignatiev A, Van Hove MA (1978) Phys Rev B 17:1510
Kitakami O, Sato H, Shimada Y, Sato F, Tanaka M (1997) Phys Rev B 56:13849
Houska CR, Averbach BL, Cohen M (1960) Acta Metall 8:81
Liu J-X, Su H-Y, Sun D-P, Zhang B-Y, Li W-X (2013) J Am Chem Soc 135:16284
Zhuo MK, Tan KF, Borgna A, Saeys M (2009) J Phys Chem C 113:8357
Liu JX, Su HY, Li WX (2013) Catal Today 215:36
Cheng J, Gong XQ, Hu P, Lok CM, Ellis P, French S (2008) J Catal 254:285
Zhuo MK, Borgna A, Saeys M (2013) J Catal 297:217
Ge QF, Neurock M (2006) J Phys Chem B 110:15368
Huo C-F, Li Y-W, Wang J, Jiao H (2008) J Phys Chem C 112:3840
Cheng J, Hu P, Ellis P, French S, Kelly G, Lok CM (2008) J Phys Chem C 112:9464
Cheng J, Song T, Hu P, Lok CM, Ellis P, French S (2008) J Catal 255:20
Mattsson AE, Schultz PA, Desjarlais MP, Mattsson TR, Leung K (2005) Modell Simul Mater Sci Eng 13:R1
Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ et al (1992) Phys Rev B 46:6671
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
Mason SE, Grinberg I, Rappe AM (2004) Phys Rev B 69:161401
Abild-Pedersen F, Andersson MP (2007) Surf Sci 601:1747
Bridge ME, Comrie CM, Lambert RM (1977) Surf Sci 67:393
Papp H (1983) Surf Sci 129:205
Pick S (2007) Surf Sci 601:5571
Huo CF, Li YW, Wang JG, Jiao HJ (2008) J Phys Chem C 112:14108
Inderwildi OR, Jenkins SJ, King DA (2008) J Phys Chem C 112:1305
Storsæter S, Chen D, Holmen A (2006) Surf Sci 600:2051
van Helden P, van den Berg J-A, Ciobica IM (2012) Catal Sci Technol 2:491
Gnanamani MK, Jacobs G, Shafer WD, Davis BH (2013) Catal Today 215:13
Karaca H, Safonova OV, Chambrey S, Fongarland P, Roussel P, Griboval-Constant A et al (2011) J Catal 277:14
Tan KF, Xu J, Chang J, Borgna A, Saeys M (2010) J Catal 274:121
Schulz H (2014) Catal Today 228:113
Braconnier L, Landrivon E, Clemencon I, Legens C, Diehl F, Schuurman Y (2013) Catal Today 215:18
O’Shea V, Homs N, Fierro JLG, de la Piscina PR (2006) Catal Today 114:422
Enache DI, Rebours B, Roy-Auberger M, Revel R (2002) J Catal 205:346
Ducreux O, Lynch J, Rebours B, Roy M, Chaumette P. (1998).In: Parmaliana A, Sanfilippo D, Frusteri F, Vaccari A, Arena F, editor. Natural Gas Conversion V, p. 125
Tuxen A, Carenco S, Chintapalli M, Chuang CH, Escudero C, Pach E et al (2013) J Am Chem Soc 135:2273
Zhao Y-H, Sun K, Ma X, Liu J, Sun D, Su H-Y et al (2011) Angew Chem Int Ed 50:5335
Cheng J, Hu P, Ellis P, French S, Kelly G, Lok CM (2010) J Phys Chem C 114:1085
van Dijk HAJ, Hoebink J, Schouten JC (2003) Top Catal 26:163
van Dijk HAJ, Hoebink J, Schouten JC (2003) Top Catal 26:111
van Dijk HAJ, Hoebink JHB, Schouten JC (2001) Chem Eng Sci 56:1211
Kruse N, Schweicher J, Bundhoo A, Frennet A, de Bocarme TV (2008) Top Catal 48:145
Frennet A, de Bocarme TV, Bastin JM, Kruse N (2005) J Phys Chem B 109:2350
Acknowledgments
The financial support from The Norwegian Research Council through the NFR 209337 Programme is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Qi, Y., Yang, J., Chen, D. et al. Recent Progresses in Understanding of Co-Based Fischer–Tropsch Catalysis by Means of Transient Kinetic Studies and Theoretical Analysis. Catal Lett 145, 145–161 (2015). https://doi.org/10.1007/s10562-014-1419-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-014-1419-x