
Abstract
Introduction
Duchenne muscular dystrophy (DMD) is induced by a wide spectrum of mutations such as exon deletions, duplications and small mutations in the dystrophin gene. This is the first study on the mutational spectrum in a cohort of DMD children from India, with an emphasis to compare the mutations in familial and sporadic forms.
Results
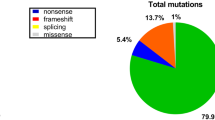
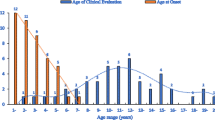
Multiplex ligation-dependent probe amplification (MLPA) and next-generation sequencing (NGS) identified 525 and 70 cases of DMD, respectively, while 11 cases showed absent dystrophin staining with no mutations detected. Families with two or more affected males contributed to 12% of the entire cohort. The mutations comprised of exonic deletions in 492/606 (81.2%), duplications in 33/606 (5.4%) and small mutations (point mutations and INDELs) in 70/606 (11.5%) cases. MLPA identified significantly more larger mutations in sporadic (88.2%) than in familial cases (75.3%). The mutations in NGS were: [nonsense = 40 (57.1%); frameshift = 17 (24.3%); splice variant = 12 (17.1%)]. Nonsense mutations were more common in familial than in sporadic cases: 17.8% vs 10.7%. The familial group reported an earlier onset of disease (2.8 ± 1.7 years) as compared to sporadic cases (3.8 ± 1.6 years).
Conclusion
MLPA could identify mutations in a high percentage of our DMD children. The preponderance of small mutations was noted to be distinctly higher in the familial group. Intriguingly, the familial form of DMD formed a small percentage of the entire cohort. The reasons could be increasing awareness among parents and physicians with early identification of DMD cases, genetic counseling and prenatal testing.




Similar content being viewed by others

References
Aartsma-Rus A, Van Deutekom JCT, Fokkema IF et al (2006) Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34:135–144. https://doi.org/10.1002/mus.20586
Tennyson CN, Klamut HJ, Worton RG (1995) The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet 9:184–190. https://doi.org/10.1038/ng0295-184
Koenig M, Beggs AH, Moyer M et al (1989) The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet 45:498–506
Aartsma-Rus A, Ginjaar IB, Bushby K (2016) The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet 53:145–151. https://doi.org/10.1136/jmedgenet-2015-103387
Lalic T, Vossen RHAM, Coffa J et al (2005) Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet 13:1231–1234. https://doi.org/10.1038/sj.ejhg.5201465
Hegde MR, Chin ELH, Mulle JG et al (2008) Microarray-based mutation detection in the dystrophin gene. Hum Mutat 29:1091–1099. https://doi.org/10.1002/humu.20831
Gatta V, Scarciolla O, Gaspari AR et al (2005) Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA). Hum Genet 117:92–98. https://doi.org/10.1007/s00439-005-1270-7
Itto AB, Hamzi K, Bellayou H et al (2013) Evolution of molecular diagnosis of Duchenne muscular dystrophy. J Mol Neurosci MN 50:314–316. https://doi.org/10.1007/s12031-013-9971-1
Tuffery-Giraud S, Béroud C, Leturcq F et al (2009) Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat 30:934–945. https://doi.org/10.1002/humu.20976
Bladen CL, Salgado D, Monges S et al (2015) The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 36:395–402. https://doi.org/10.1002/humu.22758
Flanigan KM, Dunn DM, von Niederhausern A et al (2009) Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 30:1657–1666. https://doi.org/10.1002/humu.21114
Okubo M, Goto K, Komaki H et al (2017) Comprehensive analysis for genetic diagnosis of Dystrophinopathies in Japan. Orphanet J Rare Dis 12:149. https://doi.org/10.1186/s13023-017-0703-4
Yang J, Li SY, Li YQ et al (2013) MLPA-based genotype–phenotype analysis in 1053 Chinese patients with DMD/BMD. BMC Med Genet 14:29. https://doi.org/10.1186/1471-2350-14-29
Swaminathan B, Shubha GN, Shubha D et al (2009) Duchenne muscular dystrophy: a clinical, histopathological and genetic study at a neurology tertiary care center in Southern India. Neurol India 57:734–738. https://doi.org/10.4103/0028-3886.59468
Manjunath M, Kiran P, Preethish-Kumar V et al (2015) A comparative study of mPCR, MLPA, and muscle biopsy results in a cohort of children with Duchenne muscular dystrophy: a first study. Neurol India 63:58. https://doi.org/10.4103/0028-3886.152635
Vengalil S, Preethish-Kumar V, Polavarapu K et al (2017) Duchenne muscular dystrophy and becker muscular dystrophy confirmed by multiplex ligation-dependent probe amplification: genotype–phenotype correlation in a large cohort. J Clin Neurol Seoul Korea 13:91–97. https://doi.org/10.3988/jcn.2017.13.1.91
Polavarapu K, Manjunath M, Preethish-Kumar V et al (2016) Muscle MRI in Duchenne muscular dystrophy: evidence of a distinctive pattern. Neuromuscul Disord 26:768–774
Singh R-J, Manjunath M, Preethish-Kumar V et al (2018) Natural history of a cohort of Duchenne muscular dystrophy children seen between 1998 and 2014: an observational study from South India. Neurol India 66:77
Lee T, Takeshima Y, Kusunoki N et al (2014) Differences in carrier frequency between mothers of Duchenne and Becker muscular dystrophy patients. J Hum Genet 59:46–50. https://doi.org/10.1038/jhg.2013.119
Flanigan KM (2014) Duchenne and Becker muscular dystrophies. Neurol Clin 32:671–688. https://doi.org/10.1016/j.ncl.2014.05.002 (viii)
van Ruiten HJA, Straub V, Bushby K, Guglieri M (2014) Improving recognition of Duchenne muscular dystrophy: a retrospective case note review. Arch Dis Child 99:1074–1077. https://doi.org/10.1136/archdischild-2014-306366
Magri F, Govoni A, D’Angelo MG et al (2011) Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol 258:1610–1623. https://doi.org/10.1007/s00415-011-5979-z
Wang D-N, Wang Z-Q, Yan L et al (2017) Clinical and mutational characteristics of Duchenne muscular dystrophy patients based on a comprehensive database in South China. Neuromuscul Disord NMD 27:715–722. https://doi.org/10.1016/j.nmd.2017.02.010
Liang W-C, Wang C-H, Chou P-C et al (2018) The natural history of the patients with Duchenne muscular dystrophy in Taiwan: a medical center experience. Pediatr Neonatol 59:176–183. https://doi.org/10.1016/j.pedneo.2017.02.004
Maheshwari M, Vijaya R, Kabra M et al (2000) Prenatal diagnosis of Duchenne muscular dystrophy. Natl Med J India 13:129–131
Verma PK, Dalal A, Mittal B, Phadke SR (2012) Utility of MLPA in mutation analysis and carrier detection for Duchenne muscular dystrophy. Indian J Hum Genet 18:91–94. https://doi.org/10.4103/0971-6866.96667
Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ et al (2015) DMD mutations in 576 dystrophinopathy families: a step forward in genotype–phenotype correlations. PLoS One 10:e0135189. https://doi.org/10.1371/journal.pone.0135189
Vieitez I, Gallano P, González-Quereda L et al (2017) Espectro mutacional de la distrofia muscular de Duchenne en España: estudio de 284 casos. Neurología 32:377–385. https://doi.org/10.1016/j.nrl.2015.12.009
Flanigan KM, Dunn DM, von Niederhausern A et al (2011) Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene. Hum Mutat 32:299–308. https://doi.org/10.1002/humu.21426
Crone M, Mah JK (2018) Current and emerging therapies for duchenne muscular dystrophy. Curr Treat Options Neurol 20:31. https://doi.org/10.1007/s11940-018-0513-6
Aartsma-Rus A, Fokkema I, Verschuuren J et al (2009) Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat 30:293–299. https://doi.org/10.1002/humu.20918
McDonald CM, Campbell C, Torricelli RE et al (2017) Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Lond Engl 390:1489–1498. https://doi.org/10.1016/S0140-6736(17)31611-2
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors have no conflict of interest.
Ethical standards
Approval from ‘Institutional Ethics Committee’ has been obtained.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Polavarapu, K., Preethish-Kumar, V., Sekar, D. et al. Mutation pattern in 606 Duchenne muscular dystrophy children with a comparison between familial and non-familial forms: a study in an Indian large single-center cohort. J Neurol 266, 2177–2185 (2019). https://doi.org/10.1007/s00415-019-09380-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09380-3