
Abstract
In microorganisms, the enzyme acetate kinase (AK) catalyses the formation of ATP from ADP by de-phosphorylation of acetyl phosphate into acetic acid. A mutant strain of Clostridium acetobutylicum lacking acetate kinase activity is expected to have reduced acetate and acetone production compared to the wild type. In this work, a C. acetobutylicum mutant strain with a selectively disrupted ack gene, encoding AK, was constructed and genetically and physiologically characterized. The ack − strain showed a reduction in acetate kinase activity of more than 97% compared to the wild type. The fermentation profiles of the ack − and wild-type strain were compared using two different fermentation media, CGM and CM1. The latter contains acetate and has a higher iron and magnesium content than CGM. In general, fermentations by the mutant strain showed a clear shift in the timing of peak acetate production relative to butyrate and had increased acid uptake after the onset of solvent formation. Specifically, in acetate containing CM1 medium, acetate production was reduced by more than 80% compared to the wild type under the same conditions, but both strains produced similar final amounts of solvents. Fermentations in CGM showed similar peak acetate and butyrate levels, but increased acetoin (60%), ethanol (63%) and butanol (16%) production and reduced lactate (−50%) formation by the mutant compared to the wild type. These findings are in agreement with the proposed regulatory function of butyryl phosphate as opposed to acetyl phosphate in the metabolic switch of solventogenic clostridia.
Similar content being viewed by others
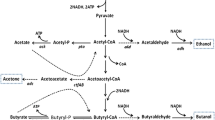

Introduction
The expected shortage of petroleum in the near future and concerns regarding the net increase of carbon dioxide emissions by fossil fuel combustion have resulted in a search for sustainable sources for the production of transport fuels and chemicals. Butanol derived from lignocellulosic materials could provide such an alternative method to the current petrochemical production process (Dürre 2008; Lee et al. 2008). Fermentative butanol production is carried out by various clostridial species as part of the acetone–butanol–ethanol (ABE) process (Green 2011; Jones and Woods 1986; Lee et al. 2008; Lütke-Eversloh and Bahl 2011). Two of the most important drawbacks of the ABE process are the low butanol yields, due to the formation of by-products and the toxicity of butanol itself to the cultures, and the high costs of the separation of the different products (Jones 2001).
Several strategies to increase the production of butanol and reduce by-product formation by metabolic engineering have been described (López-Contreras et al. 2010; Rogers et al. 2006). To date, only two gene knockouts (phosphotransacetylase, pta; butyrate kinase, buk1) (Green et al. 1996; Shao et al. 2007) and five asRNA constructs (acetoacetyl-CoA:acetate/butyrate:CoA-transferase, ctfA and ctfB; acetoacetate decarboxylase, adc; phosphotransbutyrylase, ptb; buk1) (Desai and Papoutsakis 1999; Tummala et al. 2003a, b) pertaining directly to the reduction of by-product formation in C. acetobutylicum ATCC 824 have been reported. Inactivation of buk1 resulted in a 42% increase in butanol levels (Harris et al. 2000) while inactivation of the pta, involved in acetate formation, did not show improved solvent production (WT A-B-E production, 72:133:13 mM vs. 79:131:11 for the pta − mutant) (Green et al. 1996).
In related strains, three genes have been knocked out: adc in C. acetobutylicum EA 2018 (Jiang et al. 2009) and acetate kinase (ack) and buk1, both in C. acetobutylicum M5 (Sillers et al. 2008). Impairment of the acetone pathway results in all cases in increased acid accumulation, as expected for strains that can no longer efficiently take-up acids from the growth medium. Strain M5 is a degenerated strain, isolated after chemical mutagenesis, which no longer has the pSOL1 megaplasmid and therefore is devoid of solvent production (Clark et al. 1989). To restore solvent productivity, Nair and Papoutsakis (1994) expressed the alcohol dehydrogenase gene (adhE), normally located on pSOL1, in strain M5 from a replicative plasmid. Butanol and ethanol production was restored without acetone formation, but at reduced levels compared to the wild-type ATCC 824 strain, while large amounts of acetate and butyrate accumulated in the growth medium. To reduce the acid production in this M5 background, Sillers et al. (2008) created an acetate kinase knockout strain (M5 AKKO) and a butyrate kinase knockout strain (M5 BKKO). Both strains had reduced, but not eliminated, acid accumulation corresponding to the inactivated pathway and grew more slowly. Attempts to restore solvent formation by these acid production mutants by plasmid based adhE expression were unsuccessful for the M5 BKKO strain. M5 AKKO could be transformed, but it still produced large amounts of acetate and butyrate with reduced alcohol production compared to the M5 adhE expressing strain.
Based on the fermentation results of the pta − and buk − mutants and a later study measuring the intracellular levels of phosphorylated acids in C. acetobutylicum, it was proposed that butyryl phosphate is a regulatory molecule involved in the transition from acidogenesis into solventogenesis (Zhao et al. 2005). The enzymes involved in acetate production in C. acetobutylicum, phosphotransacetylase and acetate kinase, are encoded by the pta-ack operon. A C. acetobutylicum strain in which the acetate kinase (ack) gene is inactivated is expected to accumulate acetyl phosphate, a phosphorylated intermediate with a potential regulatory function in Escherichia coli (Klein et al. 2007; McCleary et al. 1993). On the contrary, a pta − mutant is not expected to accumulate this intermediate (McCleary et al. 1993). The influence of ack disruption on solvent production in a wild-type background has not been studied yet.
Formation of acetate, butyrate, ethanol and acetone limits the amount of metabolic precursors available for butanol production. In our current research, we therefore focus on reducing formation of these by-products by a targeted gene disruption. By inactivating the genes involved in their production, we wished to investigate the effects on the overall metabolism and butanol production.
In this study, we describe a mutant of C. acetobutylicum with an inactivated acetate kinase (ack) gene, obtained using the ClosTron system for selective gene inactivation in Clostridia (Heap et al. 2007). The resulting ack − mutant has been characterized, and its fermentation profile under batch conditions has been compared to that of the wild-type strain.
Materials and methods
Bacterial strains and plasmids
All bacterial strains and plasmids used in this study are listed in Table 1. Stock cultures of C. acetobutylicum were maintained as spore suspensions in sterile 15% (v/v) glycerol at −20 °C. Chemical competent E. coli TOP 10 cells (Invitrogen) were used for vector maintenance and cloning.
Media and growth conditions
E. coli strains were grown in lysogeny broth (LB) medium (Bertani 2004; Sambrook et al. 1989) at 37 °C, 200 rpm supplemented with the appropriate antibiotics. Clostridial spore suspensions were prepared as previously described (Siemerink et al. 2011) and heat-shocked for 10 min at 70 °C, prior to using them as an inoculum for pre-cultures. Liquid cultures of C. acetobutylicum were grown in CM1 medium, based on Nimcevic et al. (1998), containing per litre: yeast extract, 5.00 g; KH2PO4, 1.00 g, K2HPO4, 0.76 g; ammonium acetate, 3.00 g; para-aminobenzoic acid (pABA), 0.10 g; MgSO4•7 H2O, 1.00 g; FeSO4•7 H2O, 0.50 g and glucose, 100 g or in CGM (Roos et al. 1985) containing per litre: yeast extract, 5.00 g; KH2PO4, 0.75 g; K2HPO4, 0.75 g; asparagine•H2O, 2.27 g; (NH4)2SO4, 2.00 g; cysteine, 0.50 g; MgSO4•7 H2O, 0.40 g; MnSO4•H2O, 0.01 g; FeSO4•7 H2O, 0.01 g and glucose, 80 g. Media were made anaerobic by flushing with nitrogen gas for 10 to 30 min depending on the volume of the liquid. All clostridial culture experiments were performed at 37 °C, without shaking, and anaerobically in (a) an anaerobic chamber (Sheldon Manufacturing, Oregon USA; gas mixture consisting of 15% CO2, 4% H2 and 81% N2) or (b) in glass serum vials as described previously (López-Contreras et al. 2000).
Culture media were supplemented with ampicillin (100 μg mL-1), chloramphenicol (30 μg mL-1), thiamphenicol (15 μg mL-1), erythromycin (40 μg mL-1 standard; 5–10 μg mL-1 for initial mutant isolation) or kanamycin (50 μg mL-1) when appropriate. Biomass was determined spectrophotometrically (Pharmacia Biotech Ultrospec 2000) based on a experimentally determined relationship between optical density measurements (OD600) and cell dry weight [CDW = (OD600–0.40)/2.91].
DNA isolation, manipulation and transformation
Standard molecular work was done according to established protocols (Sambrook et al. 1989). DNA from C. acetobutylicum was isolated using the GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich) using the Gram-positive isolation procedure which incorporates a lysozyme (from chicken egg white; Fluka) treatment. E. coli plasmid DNA was isolated by the GenElute Plasmid Miniprep Kit (Sigma-Aldrich). Both kits were used according to the manufacturers’ instructions. DNA amplification by PCR on C. acetobutylicum DNA was done using Pwo polymerase (Roche Diagnostics), and E. coli colony PCR reactions were carried out using REDTaq DNA polymerase (Sigma-Aldrich).
Prior to transformation into C. acetobutylicum, plasmids were methylated in vivo (Mermelstein and Papoutsakis 1993) by electroporation into E. coli TOP 10 (pAN2) cells. Electrotransformation of C. acetobutylicum was carried out as previously described (Oultram et al. 1988). All manipulations were carried out anaerobically and on ice. After transformation and recovery, the cells were plated on pre-warmed plates containing thiamphenicol.
Primers and DNA sequencing
All DNA primers used in the study are listed in Table 2. Primers were obtained from Eurogentec (Seraing, Belgium). DNA sequencing of clones was done by BaseClear (Leiden, the Netherlands).
Construction of ClosTron plasmids
Plasmids were constructed according to the protocol by Heap et al. (2007) using the Sigma Targetron design website (www.sigma-genosys.com/targetron/). The retargeted cassettes were ligated into the pMTL007 backbone, resulting in plasmids pMTL007::Cac-ack-1027a and pMTL007::Cac-ack-84 s. Correct plasmids were identified by restriction digestion and by sequencing the retargeted region using primers 5402F_fwd and pMTL007_rev.
Induction of the ClosTron system and mutant isolation and verification
One-millilitre CGM with thiamphenicol was inoculated with a 100-μL stationary phase overnight culture and incubated for 1.5 h at 37 °C. Then IPTG was added to a final concentration of 1 mM, and incubation was continued for 3 h. Cells were centrifuged at 5,200 × g for 1 min, and the supernatant was removed. After addition of 0.5 mL PBS, cells were resuspended and centrifuged again at 5,200 × g for 2 min. The supernatant was removed and replaced with 1-mL CGM without antibiotics. The cells were resuspended and incubated for 3 h at 37 °C before plating on CGM agar plates containing 5 μg mL−1 erythromycin and incubated for 1 or 2 days at 37 °C. Selected colonies were restreaked on plates containing 10 μg mL−1 erythromycin. Isolated mutants were checked for absence of the 1.3-kb plasmid sized, intron I containing, ermB gene using primers ErmRAM-F and ErmRAM-R.
Southern blot
Genomic DNA of the wild type, the AK mutant and the pMTL007 plasmid was restriction digested using four different mixes for each sample (KpnI and SacI, BglII and EcoRV, HindIII, HpaII). Digests were then subjected to Southern blot analysis using a random-labelled DIG probe (Roche) and was performed according to the manufacturer’s instructions. Primers TT_Probe_f and TT_Probe_r were used to generate the probe, with pMTL007 plasmid DNA as a template. The resulting probe hybridised to nucleotides 312 to 694 of the inserted intron II sequence. Irrespective of restriction enzyme used, no DNA fragment was released from the wild-type genomic DNA that hybridised to the probe. In contrast, restriction fragments of the expected size were derived from both the genome of the AK mutant and the ClosTron plasmid which gave a positive signal on the Southern blot.
Cell-free extracts and enzymatic activity assays
C. acetobutylicum cells were harvested from cultures with an OD of approximately 1.0 by centrifugation (15,000 × g, 7 min, 4 °C) and resuspended in 50 mM MOPS buffer (pH = 7.0) containing 1 mM DTT. Crude cell extracts were prepared by French press (Thermo Scientific) homogenization (two passes at 16,000 psi). The homogenates were centrifuged (20,817 × g, 15 min, 4 °C) decantated and centrifuged as before. The cell-free extracts were stored at −80 °C until analysed. AK and BK were assayed at 29 °C in the acyl phosphate forming direction according to the method of Rose (1955). Units of AK or BK activity were defined as micromoles of substrate converted per minute. Acetyl phosphate was used for preparation of a calibration curve for both reactions. Phosphotransacetylase (PTA) and phosphotransbutyrylase (PTB) activities were measured in the acyl phosphate forming direction by measuring the release of coenzyme A as previously described (Cary et al. 1988). Units of PTA or PTB activity were defined as micromoles of CoA released per minute. Homogenisation and enzyme assays were carried out aerobically. Total protein in crude extracts was determined using Quick Start Bradford Protein Assay (Bio-Rad) with bovine serum albumin as a standard.
Batch fermentations of C. acetobutylicum
Fermentations were performed in 2 L bioreactors (1 L working volume) controlled via a Bio Controller ADI 1010 by a PC running Bioexpert software (all Applikon, the Netherlands). No antibiotics were used during the fermentations. CGM batch fermentations were set up as follows: Yeast extract, phosphates, ammonium sulphate and asparagine were autoclaved with the reactor, whereas autoclaved glucose was added to the reactor after cooling. Metal sulphates and cysteine were added filter-sterilised. CM1 medium-based batch fermentations were setup by autoclaving yeast extract, phosphates and ammonium acetate with the reactor. Autoclaved glucose was added to the reactor after cooling. Metal sulphates and pABA were added as a filter sterilised mix.
The medium in the reactor was sparged overnight with nitrogen. Prior to inoculation, Sigma antifoam 204 was added to the reactor (0.1‰). The reactor was inoculated with 50 mL of an overnight pre-culture grown on the same medium but supplemented with erythromycin in the case of the mutant. After inoculation, the starting pH was allowed to fall to 5.0, after which it was controlled by addition of 6 M ammonium hydroxide solution (CGM) or 4 M KOH (CM1). During the fermentation, the headspace was flushed with nitrogen and the outgoing gas flow was passed through a condenser at 4 °C. The agitation rate was 200 rpm.
Analysis of metabolites
Metabolites present in culture supernatants (glucose, acetate, butyrate, lactate, acetoin, meso-2,3-butanediol, acetone, butanol and ethanol) were determined by HPLC as described previously (Siemerink et al. 2011) using valeric acid as an internal standard.
Results
ack gene disruption
Sequencing of the acetate kinase gene (ack or askA, CA_C1743) of the C. acetobutylicum WUR strain showed that it is identical to the published genomic sequence of the ATCC 824 strain (data not shown). After submitting the gene sequence to the TargeTron website, ten possible insertion sites were returned. The two top scoring ones were selected for our mutagenesis work. Each site requires its own set of three unique primers and a general one (EBS_Univ) for the splicing by overlap extension (SOEing) PCR (Heap et al. 2007; Ho et al. 1989) that is used to replace the standard sequence with a sequence that will recognise the desired insertion site. The first set of unique primers, consisting of AK1027a_ibs, AK1027a_ebs1d and AK1027a_ebs2, was used to target the intron to insert in the anti-sense orientation between base pairs 1027 and 1028 of ack. The second set of unique primers, consisting of AK84s_ibs, AK84s_ebs1d and AK84s_ebs2, targeted the intron to insert in the sense orientation between base pairs 84 and 85 (Table 2).
The retargeted plasmids were constructed as described in the materials and methods section. Transformation of the plasmids, selection and induction of the intron II system resulted in erythromycin-resistant colonies. Colonies were screened for insertion in the ack gene by PCR on genomic DNA using primers Cac_ack_fwd and Cac_ack_rev. Of the eight colonies screened, three were positive for insertion between base pairs 1027 and 1028. Of the 16 screened colonies that contained the pMTL007::Cac-ack-84 s construct, none showed insertion in the ack gene. All of the screened colonies tested positive by PCR for a correctly sized ermB gene of approximately 900 bps (the length of the active, genomically inserted, ermB gene). This shows that in the cases where the ack gene was not affected, the intron II must have integrated somewhere else in the genome.
The insertion efficiency at the intended site of the pMTL007::Cac-ack-84 s system appeared to be lower than that for the pMTL007::Cac-ack-1027a system, and therefore, no further attempts were made to isolate a mutant with insertion at the 84/85 site. One of the successful 1027a insertants was selected and further purified by re-streaking on fresh erythromycin plates. DNA isolated from this mutant strain was analysed by PCR amplification of the full-length ack gene using primers Cac_ack_fwd and Cac_ack_rev. Only a 3.0-kbp PCR product was obtained, corresponding to the ack gene with an inserted intron. The absence of a wild type, 1.2 kbp amplicon, indicated that a pure culture was obtained (Fig. S1). Loss of the pMTL007 plasmid by the mutant was confirmed by PCR using the ermB gene primers (not shown). To check if only one copy of the intron had inserted in the genome, a Southern blot was performed. The probe employed was complementary to nucleotides 312 to 694 of the inserted intron sequence. Only bands, corresponding to the expected sizes, were detected (Fig. S2). Also this analysis confirmed that pMTL007 plasmid DNA was not present in the sample. Insertion of the intron II at the intended site in the ack gene was confirmed by sequencing (data not shown). Spore suspensions from the obtained mutant, designated C. acetobutylicum AK, were used for further studies.
Activities of acetate and butyrate pathway enzymes
The characterisation of the mutant was performed using two different media. CGM medium, which has been used previously for the characterisation of similar mutants (Green et al. 1996) and does not contain acetate, was selected in order to be able to compare our results to earlier reports and to observe acid production without background levels, while CM1 medium is used routinely in our laboratory for growth experiments and contains 39 mM acetate and a higher concentration of iron and magnesium than CGM.
To confirm functional inactivation of acetate kinase, cell-free extracts of exponentially growing cultures were assayed for enzyme activities involved in acid production. The determined specific enzyme activities are shown in Table 3. Acetate kinase enzyme activity in cells from cultures grown in both media was reduced by more than 97% compared to the wild type. The PTA activity, encoded by the pta gene which resides upstream of ack in the same operon, was also negatively affected by the insertion, being reduced by 41% in CM1 medium and 29% in CGM. Interestingly, BK activity, involved in butyrate production and encoded by the ptb–buk1 operon, also showed reduced activity in both media. While PTB activity was still comparable (90% and 100% activity of the wild type in CM1 and CGM medium, respectively), BK activity was reduced to 72% and 53% activity of the wild type in, respectively, CM1 and CGM medium.
Characterisation of the C. acetobutylicum AK mutant in batch cultures on CM1 medium
Product formation by the mutant strain was studied using pH-controlled batch cultures. The initial pH of the cultures was 6.6, and it was allowed to drop to 5.0 before it was controlled by addition of potassium hydroxide. Final product concentrations of duplicate fermentations after approximately 47 h are presented in Table 4. Figure 1 shows a typical fermentation profile in this medium. During fermentations by the wild-type strain on CM1 medium, both acetate and butyrate accumulated in the broth during the acidogenic phase of the fermentations. Butyrate production reached an average maximum of 62 mM, whereas the maximum average acetate concentration was 85 mM, a 43-mM increase relative to the starting concentration of 42 mM (Table 4). Both acids were, to some extent, taken up during the solventogenic phase of the fermentation. Butyrate levels dropped to around 16 mM, but acetate levels remained 27 mM above the starting concentration (Fig. 1a). Solvent production by the wild type reached concentrations of 161, 101 and 30 mM for butanol, acetone and ethanol, respectively. The C. acetobutylicum AK strain consumed somewhat less glucose but had increased acetate assimilation. Final concentrations of solvents of C. acetobutylicum AK strain fermentations (157, 98 and 35 mM for butanol, acetone and ethanol, respectively) were similar to those in wild-type fermentations. The production of acids by the AK mutant was markedly different. Acetate levels increased with only 9 mM, a reduction of 79% compared to the wild-type strain. The maximum butyrate levels were also lower compared to the wild-type fermentations (47 vs. 62 mM; Fig. 1c). During the solventogenic phase of fermentations by the AK strain, butyrate was re-assimilated to reach similar levels as in fermentations by the wild-type strain. Contrary to butyrate, the acetate re-assimilation by the AK mutant resulted in net consumption (7 mM) of initially present acetate.

Typical optical density and production profile of a wild-type (a and b) and AK mutant strain (c and d) fermentation in pH 5.0 controlled batch reactors using CM1 medium containing 100 g L−1 glucose. a, c Production of acids  acetate,
acetate,  butyrate,
butyrate,  lactate, solid line pH,
lactate, solid line pH,  , OD600. b, d production of solvents
, OD600. b, d production of solvents  ethanol,
ethanol,  butanol,
butanol,  acetone,
acetone,  acetoin,
acetoin,  OD600
OD600
Characterisation of the AK mutant in batch cultures on CGM medium
To allow more precise monitoring of acetate levels, we characterised the mutant in CGM medium, which contrary to CM1 does not contain any acetate. In addition, CGM medium has been used in earlier fermentation studies with C. acetobutylicum (Green et al. 1996). Figure 2 shows typical optical density and production profiles of acids and solvents from wild-type and AK strain fermentations. The average product levels for three independent fermentations per strain after a minimum of 72 h are shown in Table 4. Fermentations by the AK mutant strain consistently reached a higher biomass concentration than the wild type. Typical values of dry weight biomass concentration obtained were 4.5 ± 0.3 mg mL−1 for the wild-type and 5.5 ± 0.1 mg mL−1 for the AK mutant (Table 4). An interesting observation was that the fermentation broths from the AK mutant strain showed an intense yellow colour at the end of every fermentation, which was not the case for wild-type cultures. Spectrofotometrical analysis of the broth supernatant gave a local absorption maximum at 450 nm, suggesting increased build-up of riboflavin (vitamin B2) in the medium.

Typical optical density and production profile of a wild-type (a and b) and AK mutant strain (c and d) fermentation in pH 5.0 controlled batch reactors using CGM containing 80 g L−1 glucose. a, c Production of acids  acetate,
acetate,  butyrate,
butyrate,  lactate, solid line pH,
lactate, solid line pH,  OD600. b, d Production of solvents
OD600. b, d Production of solvents  ethanol,
ethanol,  butanol,
butanol,  acetone,
acetone,  acetoin,
acetoin,  OD600
OD600
During the fermentation, acetate was still produced by the AK strain but at slightly reduced maximum levels (WT 30 mM, AK 26 mM) compared to the wild type, as is shown in Fig. 2a, c. Maximum butyrate levels also did not seem to be affected to a great extent, with somewhat higher values for the mutant (WT 38 mM, AK 43 mM). There was, however, a clear difference at what time point, compared to one another, the maximum acetate and butyrate levels were reached. The production of acetate by the AK mutant was delayed compared to that of butyrate. This difference was best observed by plotting the ratio between butyrate and acetate at various time points (Fig. 3a). From the start of the fermentation by the wild type, the ratio between butyrate and acetate rose, levelling off after 9 h at 1.5 and stayed at that ratio for 7 h. The ratio in fermentations by the AK strain continued to increase with a maximum of 4.3 after 14 h. The ratio then decreased to similar levels as those observed for the wild type, and both reached a minimum of 0.1. Analysis of the fermentations with CM1 medium gave a similar difference in acid ratio production (Fig. 3b).

Butyrate–acetate ratios in CGM (a) or butyrate-relative acetate ratios CM1 (b) of a wild-type strain ( ) and AK mutant strain (
) and AK mutant strain ( ) fermentation in pH 5.0 controlled batch reactors. On the horizontal axis, normalized time is plotted to account for variations in lag time of the cultures. In the graph, 9 h corresponds to an OD600 of 1.0. Relative acetate is calculated by the subtracting the initial acetate level at time of inoculation from the measured acetate levels later during the fermentation
) fermentation in pH 5.0 controlled batch reactors. On the horizontal axis, normalized time is plotted to account for variations in lag time of the cultures. In the graph, 9 h corresponds to an OD600 of 1.0. Relative acetate is calculated by the subtracting the initial acetate level at time of inoculation from the measured acetate levels later during the fermentation
Lactate, normally only detected at low levels (Green et al. 1996; Jones and Woods 1986), if at all, in C. acetobutylicum ATCC 824 fermentations in CGM medium, was produced in all three fermentations by C. acetobutylicum WUR (57 ± 7 mM). The AK mutant also produced lactate but consistently at lower levels (28 ± 2 mM). Various attempts—filter sterilisation of minerals, reduced stirring speed, extended sparging time with nitrogen gas before inoculation—were made to reduce lactate production in fermentations by the wild-type, but without success.
Butanol and ethanol production by the AK mutant were, respectively, 16% and 59% higher compared to the wild-type production. Acetone production levels were not significantly affected. The conversion of glucose to butanol by the mutant had increased with 19%, and total solvent yields from glucose were up with 21% (0.54 mol ABE/mol glucose consumed vs. 0.66 mol ABE/mol glucose consumed). The final concentration of the by-product acetoin had increased with 58%, from 15 mM with the wild type to 24 mM for the mutant.
Discussion
The inactivation of the butyrate kinase (buk1) gene of C. acetobutylicum has previously been shown to result in reduced butyrate formation, increased peak acetate levels and increased butanol and ethanol production (Harris et al. 2000). Acetone is one of the major by-products of butanol production as it is formed as a consequence of the uptake of acids from the medium. It has been shown through a computational approach that even under solvent forming conditions acetate production continues, while extracellular acetate levels drop (Desai et al. 1999). Produced acetate is continuously converted to acetyl-CoA, mediated by the enzyme acetoacetyl-CoA:acetate/butyrate:CoA transferase, ultimately resulting in acetone production. This approach allows C. acetobutylicum to continue acetate-mediated ATP generation during the solventogenic phase. With the aim to reduce the formation of acetate and indirectly that of acetone, by C. acetobutylicum, we have inactivated the acetate kinase (ack) gene which encodes the enzyme catalysing the last step in acetate production. In this substrate level phosphorylation reaction, the phosphate group of acetyl phosphate is transferred to ADP, generating ATP and acetate. In addition, we were interested to see if solvent formation would be similarly beneficially affected, as reported for buk1 gene disruption (Harris et al. 2000).
In this study, site-directed gene disruption was accomplished by integration of a group II intron using the ClosTron system (Heap et al. 2007). This system is markedly different from the non-replicative plasmid integration-based approach used for the previously generated buk1 and pta mutants of C. acetobutylicum (Green et al. 1996). Mutations based on single cross-over recombination are inherently unstable, whereas insertion of the group II intron in the anti-sense orientation, relative to the gene, results in permanent inactivation (Heap et al. 2007). The ClosTron and a similarly adapted TargeTron system (Shao et al. 2007) have been used to generate various mutants, including adc and hbd knockout mutants (Jiang et al. 2009; Lehmann and Lütke-Eversloh 2011).
Successful insertion of the intron after transformation of pMTL007::Cac-ack-1027a would, after translation of the mutated gene, truncate the native sequence of the AK protein at amino acid 342. However, additional intron sequence encoded amino acids will be added to the native sequence. In the case of insertion after nucleotide 1027, the sequence would be extended by 86 amino acids, so intron insertion actually results in a protein that is 27 amino acids longer than the native enzyme.
Although the retrohoming system of the group II introns is expected to be site-specific, it is possible that no or only a limited number of correctly positioned insertants can be found (Heap et al. 2007; Perutka et al. 2004). Our results show efficient insertion into insertion site 1027a, but for insertion site 84 s, 16 screened colonies did not yield a correct insertant. These results emphasise the need to test multiple insertion sites when using TargeTron-based systems.
AK activity in the AK mutant was less than 3% of the wild-type enzyme activity (Table 3), demonstrating that while the insertion was close to the 3′ side of the ack gene, it still resulted in functional inactivation of the enzyme. A study by Singh-Wissmann et al. (1998) showed that a conserved glutamate residue in sequence of the AK of Methanosarcina thermophila near the C terminus is essential for enzyme activity. This glutamate is conserved in acetate kinase sequences from widely divergent organisms and is also present in the C. acetobutylicum AK (Singh-Wissmann et al. 1998). In our AK strain, this glutamate residue is not present anymore, so no activity from the enzyme encoded by the disrupted gene is expected, which is in line with the low enzyme activities observed (Table 3).
It was expected that disruption of acetate kinase would result in decreased acetate levels and increased butyrate levels to compensate for the loss in ATP production (Lee et al. 2009), but this was not immediately observed. In both tested media, the AK mutant strain had peak butyrate levels that were lower than those for the wild type, while peak acetate levels were reduced compared to wild-type fermentations. Final acetate levels were equal (CGM), or reduced in comparison to wild-type fermentations.
The fact that inactivation of AK does not abolish acetate production has been previously observed in both Clostridium tyrobutyricum (Liu et al. 2006) and the degenerated C. acetobutylicum strain M5 (Sillers et al. 2008). In the C. tyrobutyricum ack − strain, the fermentation time was extended and both acetate and butyrate levels surpassed those of the wild type. The M5 ack − strain (M5 AKKO) produced acetate at reduced levels but with similar butyrate formation. When a plasmid-based alcohol dehydrogenase (aad) is expressed in M5 AKKO, acetate production is again high (180 mM) while butyrate production levels are depended on the fermentation pH ranging from 75 mM for pH 5.5 to 287 mM for pH 6.0 fermentations (Sillers et al. 2008). Lee et al. (2009) suggest that an alternative acetate-producing pathway is operational in the M5 AKKO mutant. This could also be applicable to our WUR strain and derived mutants.
Green et al. (1996) reported on another acetate pathway mutant in C. acetobutylicum in which the pta gene was inactivated. They reported reduced acetate levels and increased butyrate levels compared to the WT under conditions favouring acid formation (pH 5.5). Later data from Zhao et al. (2005) from static flask batch cultures showed that acetate levels were reduced by approximately 50%, while butyrate levels were unaffected under these conditions. Both culturing conditions are different from those employed in this study and complicate a direct comparison. While higher butyrate levels seem to correlate with increasing pH, the reduction in acetate is common to both. This would be in agreement with our lower butyrate levels at pH 5.0, but not with the acetate levels that we observed. Apparently an AK negative mutant is more likely to produce acetate than a mutant in which PTA is inactivated. A similar observation can be made for C. tyrobutyricum pta − and ack − mutants (Liu et al. 2006; Zhu et al. 2005) and is likely due to the operon structure in both species in which ack is located downstream of pta.
Besides an alternative acetate forming pathway, another possible explanation for the continued acetate production by the ack − mutants could be that acetate production is catalysed by BK. BK from C. acetobutylicum is active on acetate with 6% relative activity, in the acetyl phosphate-forming direction, compared to butyrate, based on in vitro data (Hartmanis 1987). Although BK activity is reduced in CGM medium (Table 3), Desai and Papoutsakis (1999) showed that even if BK activity is reduced by more the 80% in an asRNA mutant, butyrate fluxes remain unaffected and butyrate accumulates in the growth medium. Apparently the butyrate production pathway has a far greater catalytic capacity, which could possibly be used for acetate formation.
CM1 and CGM media differ mainly in the respect that the former contains more iron, magnesium and 39 mM acetate (Table S1). The marked differences between fermentation performances by the wild-type and C. acetobutylicum AK strains grown on each medium are evident from the data presented in Table 4 . In CM1 medium acetate production is strongly reduced (AK mutant 9 mM vs. WT 43 mM, both in addition to the starting concentration of 42 mM), while in CGM medium the peak acetate concentrations are quite similar (AK 26 mM, WT 30 mM). The possible effect that additional acetate in the medium has could be an indication that the intracellular metabolism involved in acetate production in the AK strain is sensitive to elevated levels of extracellular acetate. Future experiments using CM1 medium with varying, initial, concentrations of acetate can help to determine if the same maximum acetate concentration of 50 mM is reached for both lower and higher starting concentrations of acetate. This would support our current observation that acetate production is inhibited at a lower concentration (50 mM) in the AK strain than for the wild type, which accumulates more than 80 mM of acetate in CM1 medium under the same conditions. This is similar to strain ATCC 824 that accumulates up to 71 mM in CGM medium (Harris et al. 2000).
In both media, however, an important difference that was observed between the mutant and the wild-type strain was the delay in the acetate production. In the wild-type strain fermentations, acetate and butyrate production occurs with approximately 1.5 (CGM) or 1.8 times (CM1) as much butyrate formed compared to acetate. Figure 3 clearly shows that the AK mutant produces considerably more butyrate [1.86 times (CGM) and 7.2 times (CM1)] than acetate in the beginning of the fermentation, in line with the expectation that acetate production would be negatively affected by an AK disruption. It appears that when the butyrate concentration has reached its maximum level, only then acetate production is allowed to reach its full potential if no extracellular acetate is present (CGM medium). If indeed BK is involved in acetate formation, then competition of acetyl phosphate and butyryl phosphate for the available enzyme could explain the delayed acetate production.
In CGM fermentations, lactate production was seen with both the wild type and the mutant. Interestingly enough, the AK mutant strain made considerably less lactate, but more acetoin. The increase in acetoin (9 mM), however, is not enough to account for the full reduction in lactate production (29 mM), but only for up to 62%. Production of acetoin does not require oxidation of a reducing equivalent (NADH) (Freier and Gottschalk 1987) as is needed for lactate production. This additional reduction capacity could have possibly been used for solvent formation instead. Lactate production in CGM fermentations has been reported before (approximately 70 mM) (Green et al. 1996) but only for mutants, not for the WT. It seems that in our hands, C. acetobutylicum WUR, contrary to the ATCC 824 strain, is more likely to produce lactate under these fermentation conditions in CGM, whereas it does not do so in CM1 medium. This is the second difference that we have observed, next to meso-2,3-butanediol production (Siemerink et al. 2011). A likely explanation for this behaviour could be that the WUR strain is sensitive to lower levels of iron and/or magnesium compared to the ATCC 824 strain. Both these metal ions are co-factors in various oxidoreductase enzymes. If the activity of these enzymes is compromised, then remodelling of the flow of reducing equivalents within the cell, ultimately leading to lactate formation, could be the result.
Inactivation of the acetate pathway in the AK mutant is expected to lead to accumulation of acetate-preceding intracellular intermediates, such as acetyl CoA and acetyl phosphate. These higher intracellular levels could then alter product formation of ethanol and metabolically more upstream intermediates such as lactate and acetoin (Zhu and Shimizu 2005). The increased acetoin and ethanol levels in the AK mutant CGM fermentations follow the expected trend (Table 4 ). The reduced lactate production levels of the AK mutant, relative to those by the wild-type, are not in line with this expectation. The wild-type C. acetobutylicum WUR strain showed (high) lactate production levels on CGM in contrast to strain ATCC 824 which produced none under similar conditions (Desai and Papoutsakis 1999; Harris et al. 2000). This dissimilarity could be due to a different regulation of gene expression or different enzymatic properties of the iso-enzymes involved in lactate formation in both strains.
Out rationale for disrupting AK activity was that it is expected to result in intracellular accumulation of acetyl phosphate. This accumulation could potentially be a molecular trigger (McCleary et al. 1993) resulting in an observable phenotype, akin to butyryl phosphate accumulation after buk1 disruption (Harris et al. 2000). In CGM, there was significant increase in acetoin, ethanol and butanol formation by the AK mutant; however, the fermentations of both the wild type and to a lesser extent the AK mutant were affected by lactate production. In CM1 medium, hardly any lactate was produced by both strains. The AK mutant produced at peak levels 34 mM (79%) less acetate compared to the wild-type strain, but no changes in the other fermentation products were seen. This supports the earlier observations of Green et al. (1996) and Zhao et al. (2005) that acetate pathway disruption in C. acetobutylicum does not result in altered solvent production.
Despite the absence of an effect on acetone and butanol formation in fermentations in CM1 medium, AK inactivation did result in increased acetate uptake in that medium and increased butyrate uptake in CGM fermentations (Table 4 ). Acetate production was only impaired when acetate was present in the medium (CM1), but in both CGM and CM1, an altered initial acetate/butyrate product ratio was observed showing a reduced acetate production rate. The absence of an impact on solvent production by the pta − mutant of Green et al. (1996) and our AK mutant in CM1 medium is in agreement with the prediction by Zhao et al. (2005) that levels of acetyl phosphate are not linked with induction of solvent formation. Potential other roles of acetyl phosphate cannot be excluded based on these experiments. It is of further interest to note that the accumulation of a yellow compound in the fermentation broth is likely to be riboflavin (Demain 1972; Pridham 1952) which can help to facilitate the economic competitiveness of the ABE fermentation process (Cai and Bennett 2011).
We now propose the construction of an alcohologenic strain, which is unable to produce either acetate or butyrate and subsequently will not produce acetone because there are no acids to be taken up from the medium. Ideally this strain would produce only the alcohols butanol and ethanol when grown on glucose. Construction of such a strain would require the combined knockout of both acetate kinase (ack) and butyrate kinase (buk1), so that it would no longer be able to perform the, proposed, in vivo complementation of acetate production by butyrate kinase. Such a mutant would also allow us to test whether the remaining acetate production in our AK mutant is due to complementation of the enzyme activity by butyrate kinase or if one or more alternative acetate-forming pathways operate in C. acetobutylicum. We are currently investigating this option.
References
Bertani G (2004) Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J Bacteriol 186(3):595–600
Cai X, Bennett GN (2011) Improving the Clostridium acetobutylicum butanol fermentation by engineering the strain for co-production of riboflavin. J Ind Microbiol Biotechnol 38(8):1013–1025
Cary JW, Petersen DJ, Papoutsakis ET, Bennett GN (1988) Cloning and expression of Clostridium acetobutylicum phosphotransbutyrylase and butyrate kinase genes in Escherichia coli. J Bacteriol 170(10):4613–4618
Clark SW, Bennett GN, Rudolph FB (1989) Isolation and characterization of mutants of Clostridium acetobutylicum ATCC 824 deficient in acetoacetyl-coenzyme A:acetate/butyrate:coenzyme A-transferase (EC 2.8.3.9) and in other solvent pathway enzymes. Appl Environ Microbiol 55(4):970–976
Demain AL (1972) Riboflavin oversynthesis. Annu Rev Microbiol 26:369–388
Desai RP, Papoutsakis ET (1999) Antisense RNA strategies for metabolic engineering of Clostridium acetobutylicum. Appl Environ Microbiol 65(3):936–945
Desai RP, Harris LM, Welker NE, Papoutsakis ET (1999) Metabolic flux analysis elucidates the importance of the acid-formation pathways in regulating solvent production by Clostridium acetobutylicum. Metab Eng 1(3):206
Dürre P (2008) Fermentative butanol production. Bulk chemical and biofuel. Ann N Y Acad Sci 1125(1):353–362
Freier D, Gottschalk G (1987) L(+)-lactate dehydrogenase of Clostridium acetobutylicum is activated by fructose-1,6-bisphosphate. FEMS Microbiol Lett 43(2):229–233
Green EM (2011) Fermentative production of butanol—the industrial perspective. Curr Opin Biotechnol 22(3):337–343
Green EM, Boynton ZL, Harris LM, Rudolph FB, Papoutsakis ET, Bennett GN (1996) Genetic manipulation of acid formation pathways by gene inactivation in Clostridium acetobutylicum ATCC 824. Microbiol-UK 142(8):2079–2086
Harris LM, Desai RP, Welker NE, Papoutsakis ET (2000) Characterization of recombinant strains of the Clostridium acetobutylicum butyrate kinase inactivation mutant: need for new phenomenological models for solventogenesis and butanol inhibition? Biotechnol Bioeng 67(1):1–11
Hartmanis MGN (1987) Butyrate kinase from Clostridium acetobutylicum. J Biol Chem 262(2):617–621
Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP (2007) The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Meth 70(3):452–464
Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77(1):51–59
Jiang Y, Xu C, Dong F, Yang Y, Jiang W, Yang S (2009) Disruption of the acetoacetate decarboxylase gene in solvent-producing Clostridium acetobutylicum increases the butanol ratio. Metab Eng 11(4–5):284–291
Jones DT (2001) Applied acetone-butanol fermentation. In: Bahl H, Dürre P (eds) Clostridia. Biotechnology and medical application. Wiley, Weinheim, pp 125–168
Jones DT, Woods DR (1986) Acetone-butanol fermentation revisited. Microbiol Rev 50(4):484–524
Klein AH, Shulla A, Reimann SA, Keating DH, Wolfe AJ (2007) The intracellular concentration of acetyl phosphate in Escherichia coli is sufficient for direct phosphorylation of two-component response regulators. J Bacteriol 189(15):5574–5581
Lee SY, Park JH, Jang SH, Nielsen LK, Kim J, Jung KS (2008) Fermentative butanol production by Clostridia. Biotechnol Bioeng 101(2):209–228
Lee JY, Jang YS, Lee J, Papoutsakis ET, Lee SY (2009) Metabolic engineering of Clostridium acetobutylicum M5 for highly selective butanol production. Biotechnol J 4(10):1432–1440
Lehmann D, Lütke-Eversloh T (2011) Switching Clostridium acetobutylicum to an ethanol producer by disruption of the butyrate/butanol fermentative pathway. Metab Eng 13(5):464–473
Liu X, Zhu Y, Yang S-T (2006) Construction and characterization of ack deleted mutant of Clostridium tyrobutyricum for enhanced butyric acid and hydrogen production. Biotechnol Prog 22(5):1265–1275
López-Contreras AM, Claassen PA, Mooibroek H, De Vos WM (2000) Utilisation of saccharides in extruded domestic organic waste by Clostridium acetobutylicum ATCC 824 for production of acetone, butanol and ethanol. Appl Microbiol Biotechnol 54(2):162–167
López-Contreras AM, Kuit W, Siemerink MAJ, Kengen SWM, Springer J, Claassen PAM (2010) Production of longer-chain alcohols from lignocellulosic biomass: Butanol, isopropanol and 2,3-butanediol. In: Waldron K (ed) Bioalcohol production, vol 3. Woodhead Publishing Series in Energy, Cambridge, pp 415–460
Lütke-Eversloh T, Bahl H (2011) Metabolic engineering of Clostridium acetobutylicum: recent advances to improve butanol production. Curr Opin Biotechnol 22(5):634–647
McCleary WR, Stock JB, Ninfa AJ (1993) Is acetyl phosphate a global signal in Escherichia coli? J Bacteriol 175(10):2793–2798
Mermelstein LD, Papoutsakis ET (1993) In vivo methylation in Escherichia coli by the Bacillus subtilis phage ϕ3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 59(4):1077–1081
Nair RV, Papoutsakis ET (1994) Expression of plasmid-encoded aad in Clostridium acetobutylicum M5 restores vigorous butanol production. J Bacteriol 176(18):5843–5846
Nimcevic D, Schuster M, Gapes JR (1998) Solvent production by Clostridium beijerinckii NRRL B592 growing on different potato media. Appl Microbiol Biotechnol 50(4):426–428
Oultram JD, Loughlin M, Swinfield TJ, Brehm JK, Thompson DE, Minton NP (1988) Introduction of plasmids into whole cells of Clostridium acetobutylicum by electroporation. FEMS Microbiol Lett 56(1):83–88
Perutka J, Wang WJ, Goerlitz D, Lambowitz AM (2004) Use of computer-designed group II introns to disrupt Escherichia coli DExH/D-box protein and DNA helicase genes. J Mol Biol 336(2):421–439
Pridham TG (1952) Microbial synthesis of riboflavin. Econ Bot 6(2):185–205
Rogers P, Chen J-S, Zidwick MJ (2006) Organic acid and solvent production. Part III: butanol, acetone and iso-propanol; 1,3- and 1,2-propanediol production; and 2,3-butanediol production. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) The prokaryotes: symbiotic associations, biotechnology, applied microbiology, vol 1, 3rd edn. Springer, Berlin, pp 672–755
Roos JW, Mclaughlin JK, Papoutsakis ET (1985) The effect of pH on nitrogen supply, cell lysis, and solvent production in fermentations of Clostridium acetobutylicum. Biotechnol Bioeng 27(5):681–694
Rose IA (1955) Acetate kinase of bacteria (acetokinase). Methods Enzymol 1:591–595
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor
Shao L, Hu S, Yang Y, Gu Y, Chen J, Yang Y, Jiang W, Yang S (2007) Targeted gene disruption by use of a group II intron (targetron) vector in Clostridium acetobutylicum. Cell Res 17(11):963–965
Siemerink MA, Kuit W, Lopez Contreras AM, Eggink G, van der Oost J, Kengen SW (2011) Heterologous expression of an acetoin reductase leads to d-2,3-butanediol production in Clostridium acetobutylicum. Appl Environ Microbiol 77(8):2582–2588
Sillers R, Chow A, Tracy B, Papoutsakis ET (2008) Metabolic engineering of the non-sporulating, non-solventogenic Clostridium acetobutylicum strain M5 to produce butanol without acetone demonstrate the robustness of the acid-formation pathways and the importance of the electron balance. Metab Eng 10(6):321–332
Singh-Wissmann K, Ingram-Smith C, Miles RD, Ferry JG (1998) Identification of essential glutamates in the acetate kinase from Methanosarcina thermophila. J Bacteriol 180(5):1129–1134
Tummala SB, Junne SG, Papoutsakis ET (2003a) Antisense RNA downregulation of coenzyme a transferase combined with alcohol-aldehyde dehydrogenase overexpression leads to predominantly alcohologenic Clostridium acetobutylicum fermentations. J Bacteriol 185(12):3644–3653
Tummala SB, Welker NE, Papoutsakis ET (2003b) Design of antisense RNA constructs for downregulation of the acetone formation pathway of Clostridium acetobutylicum. J Bacteriol 185(6):1923–1934
Zhao YS, Tomas CA, Rudolph FB, Papoutsakis ET, Bennett GN (2005) Intracellular butyryl phosphate and acetyl phosphate concentrations in Clostridium acetobutylicum and their implications for solvent formation. Appl Environ Microbiol 71(1):530–537
Zhu J, Shimizu K (2005) Effect of a single-gene knockout on the metabolic regulation in Escherichia coli for d-lactate production under microaerobic condition. Metab Eng 7(2):104–115
Zhu Y, Liu X, Yang ST (2005) Construction and characterization of pta gene-deleted mutant of Clostridium tyrobutyricum for enhanced butyric acid fermentation. Biotechnol Bioeng 90(2):154–166
Acknowledgements
The authors would like to thank Ms H. van der Wal for her technical support. This project is financially supported by the Netherlands Ministry of Economic Affairs and the B-Basic partner organisations (www.b-basic.nl) through B-Basic, a public–private Netherlands Organisation for Scientific Research–Advanced Chemical Technologies for Sustainability programme.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 98 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kuit, W., Minton, N.P., López-Contreras, A.M. et al. Disruption of the acetate kinase (ack) gene of Clostridium acetobutylicum results in delayed acetate production. Appl Microbiol Biotechnol 94, 729–741 (2012). https://doi.org/10.1007/s00253-011-3848-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3848-4