
Abstract
P2X receptors for ATP comprise a distinct family of ligand gated ion channels with a range of properties. They have been shown to be involved in a variety of physiological processes including blood clotting, sensory perception, pain sensation, bone formation as well as inflammation and may provide a number of novel drug targets. In addition to the orthosteric site for ATP binding it has been suggested that there may be additional allosteric sites that regulate agonist action at the receptor. There is currently no crystal structure available for P2X receptors and the lack of sequence similarity to other ATP binding proteins has meant that a mutagenesis-based approach has been used primarily to investigate receptor structure-function. This review aims to provide an overview of recent work that gives an insight into residues involved in ATP action and allosteric regulation.
Similar content being viewed by others

P2X receptors and ATP action
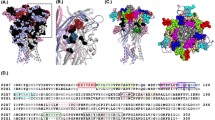
P2X receptors comprise a distinct family of ligand gated cation channels. Genes encoding seven P2X receptor subuints (P2X1–7) have been cloned and they form as homo- and heterotrimeric receptors with a range of properties (North 2002). ATP binds to the extracellular loop of the P2X receptors and it has been suggested that three molecules of agonist are required to bind and open the ion channel (Bean 1990; Ding and Sachs 1999). P2X receptors do not contain consensus sequences for agonist binding that have been identified in other ATP sensitive proteins, e.g. the Walker motif (Walker et al. 1982). This suggests that the P2X receptors have a novel ATP binding site. The extracellular loop contains ∼280 amino acids and greater than 90 of these residues are conserved in at least five of the seven mammalian P2X receptor subunits. The contribution of the majority of these conserved amino acids has been studied using alanine scanning mutagenesis. In systematic studies on the human P2X1 receptor the roles of conserved charged (Ennion et al. 2000, 2001), aromatic (Roberts and Evans 2004), structural (Ennion and Evans 2002; Digby et al. 2005; Roberts and Evans 2005) and polar amino acids (Roberts and Evans 2006) have been studied (for a review see Vial et al. 2004; Roberts et al. 2006). This work has shown that the majority of individual alanine substitutions have little or no effect on ATP potency at the receptor, however for a small number of the mutant receptors there was a large decrease in ATP sensitivity (Fig. 1). A similar picture emerges from alanine mutagenesis of conserved charged and polar residues at P2X2 receptors (Jiang et al. 2000). These studies have allowed for the generation of a model of the ATP binding site (P2X1 receptor numbering). It has been proposed that the negatively charged phosphate group of ATP is co-ordinated between positively charged lysine residues K68, K70 and K309 and the adenine ring may be sandwiched between two aromatic regions F185T186 and N290F291R292 (Roberts et al. 2006) (Fig. 2).

Summary of mutagenesis data for P2X receptors. The sequence corresponding to the extracellular domain and parts of the adjacent transmembrane domains (TM1 and TM2) of the human P2X1 receptor is shown. The ten conserved cysteine residues are shown in black. Conserved residues that when mutated to alanine or cysteine have a significant effect on ATP potency shown in orange and underlined. Purple residues correspond to where alanine substituion of conserved aromatic residues gave rise to non-functional channels. Alanine or cysteine mutants of the human P2X1 receptor that had no or <10 effect on ATP potency are shown as dashed grey lines. Residues in khaki correspond to those in other P2X receptor subunits that when mutated have an effect on regulation of ATP responses by metal ions or pH

Model of the proximity of the orthosteric and cysteine rich allosteric binding sites at P2X receptors based on mutational analysis. The putatitive orthosteric ATP binding site incorporating K68, K70, F185, N290, F291, R292 and K309 identified from mutagenesis studies based on a model of synpasin II is shown (Roberts and Evans 2007), it is likely that the residues K68 and K70 are contributed from an adjacent subunit giving rise to an inter subunit binding site (Marquez-Klakaet al. 2007). The cysteine (black circles) rich allosteric sites are shown (not to scale), the numbers correspond to the order of the conserved cysteinesin the extracellular loop with predicted disulphide bonds (indicated by black dotted lines) in the pairings 1–6, 2–4, 3–5 and 7–8. The cysteine rich regions 1–6 and 7–8 are in adjacent P2X receptor subunits as suggested by the disulphide bond (indicated by khaki dotted line) that can be formed between cysteine substitution mutants for histidines120 and 213 in the P2X2 receptor (Nagaya et al. 2005). Other residues that when mutated have effects on allosteric regulation are shown as khaki circles. The residue in pink corresponds to the arginine residues at the P2X7 receptor that can be ADP ribosylated and leads to activation of the channel (Adriouch et al. 2007), the red dotted line indicates the suggested proximity of this site to the ATP binding site
The model has been supported by two recent cysteine mutagenesis studies (Marquez-Klaka et al. 2007; Roberts and Evans 2007). Using a scanning approach the contribution of the 44 amino acids before the second transmembrane domain of the P2X1 receptor was determined (Roberts and Evans 2007). This work showed that in this region mutants N290C, F291C, R292C and K309C had reduced ATP potency and this was also reflected in a decrease in binding of radiolabelled 2-azidoATP. However it was clear from comparing the effects on the extent of the decrease in ATP potency with the effects on 2-azido ATP binding that there were likely to be effects on both agonist binding and the gating of the channel for some of these mutants. A recent study using single channel recordings from P2X2 receptor mutants has suggested that for the P2X2 receptor K308 also contributes to channel gating (Cao et al. 2007). Modification of the cysteine mutants in the P2X1 receptor with thiol reactive methanethiosulphonate reagents produced additional shifts in ATP potency consistent with a contribution of these residues to ATP binding (Roberts and Evans 2007). The fact that there was little or no change in ATP potency at the other 40 cysteine mutants studied in this region further highlights the importance of these residues in mediating ATP action.
Marquez-Klaka et al. (2007) also used a cysteine mutagenesis approach to look at the contribution of residues suggested to form the ATP binding site. They reported similar changes in ATP potency from mutants of the rat P2X1 receptor and in chimaeric rat P2X2–1 receptors. They then tested whether any of these cysteine substituted residues were close enough to form a disulphide bond. They found that K68C and F291C mutants could form a disulphide bond, and that this bond formation was inhibited by ATP (Marquez-Klaka et al. 2007) providing some molecular dimensions for the proximity of these residues. Taken together these cysteine mutagenesis studies provide strong evidence that K68, F291 and K309 are intimately involved in determining ATP action at the receptor, most likely as forming the agonist binding site.
One of the ongoing questions about the ATP binding site has been whether it is formed within or between subunits. The first experimental evidence to support that ATP action was co-ordinated between subunits was provided by Wilkinson et al. (2006). They co-expressed WT and alanine substitution mutants of the lysine residues equivalent to K68 and K309 (P2X1 receptor numbering). This work suggested that the P2X2/3 heteromeric channel contains one P2X2 and two P2X3 receptor subunits, and that residues from two different subunits interact to regulate ATP binding and/or channel gating (Wilkinson et al. 2006). The work of Marques-Klaka et al. (2007) also provides structural support for the idea that the interface between subunits is important for regulating ATP action as the disulphide bond that they showed could form between K68C and F291C was between adjacent subunits.
There was some suggestion that P2X receptors may not share a common ATP binding site as P2X4 receptors mutagenesis studies support other amino acids in the second half of the extracellular domain that are important for determining ATP sensitivity and are consistent with an agonist binding site based on sequence similarity to the family of tRNA synthetases (Stojilkovic et al. 2005; Yan et al. 2005). However more recent work has also shown that the residues predicted from the P2X1 receptor model show similar effects at P2X4 receptors (Zemkova et al. 2007). This suggests that there may be a core ATP binding site that is common to P2X receptors but there are subtype specific differences that would be consistent with the differences in pharmacological properties between the receptors.
The functional expression of a P2X receptor channel from the amoeba Dictyostelium discoideum (DdP2X) has provided some interesting insight into the modelling of the receptors (Fountain et al. 2007). Many residues that are conserved across the mammalian and Schistosome P2X receptors are absent from the DdP2X receptor (Fountain et al. 2007) however four of the seven residues proposed to be part of the mammalian P2X receptor are totally conserved and at one other position there is a conservative substitution (K70, F185, T186, K309 and F291 respectively P2X1 receptor numbering). Positive charges equivalent to K70 and K309 in the P2X1 receptor are conserved in the DdP2X receptor and when mutated to alanine result in a decrease in ATP potency as found in a range of P2X receptors (Ennion et al. 2000; Jiang et al. 2000; Wilkinson et al. 2006; Zemkova et al. 2007) consistent with the proposed role of these charged residues in co-ordinating the negatively charged phosphate of ATP. The N290F291R292 motif (P2X1 receptor numbering) that is totally conserved throughout mammalian and the Shistosoma P2X receptors is absent from DdP2X (Fountain et al. 2007). However there is a conserved tyrosine residue at an equivalent position to the phenylalanine in the whole family of DdP2Xs suggesting that there is always an aromatic amino acid at this position. It is interesting that it is the equivalent phenylalanine residue (F291) in the P2X1 receptor that has been shown to be close to K68 in an adjacent subunit by cysteine substitution and disulphide bond analysis (Marquez-Klaka et al. 2007) and that formation of this bond is blocked by ATP binding. It will be interesting to determine whether the tyrosine of the DdP2X plays an equivalent role in mediating ATP action.
The other residues F185T186 (P2X1 receptor numbering) that have been suggested to form part of the ATP binding site are also conserved in the DdP2X however the alanine replacement of the conserved phenyalanine had little or no effect on ATP sensitivity suggesting that it does not play an essential role in agonist action. At the P2X1 receptor the equivalent alanine mutation only gives rise to a modest ∼10-fold decrease in ATP potency suggesting that the aromatic residues does not make a major contribution to agonist sensitivity, however the efficacy of the partial agonists BzATP and Ap5A were essentially abolished at this mutant (Roberts and Evans 2004). This is the same as that seen for the P2X1 receptor mutants K68A, F291A, R292A and K309A (Roberts and Evans 2004) that have been widely suggested to contribute to the ATP binding site. The conserved neighbouring threonine when mutated to alanine (T186A) decreased ATP potency at the P2X1 receptors by six fold and abolished the efficacy of BzATP and Ap5A (Roberts and Evans 2006) and at P2X2 receptors the T186A mutant reduced ATP potency by ∼ 100 fold (Jiang et al. 2000). These mammalian studies suggest that the F185T186 region plays a role in the regulation of agonist action at P2X receptors although it remains to be determined directly whether this results from an effect on agonist binding or gating of the channel. The effects of the partial agonists on the equivalent F-A and T-A mutants in receptors other than P2X1 have yet to be determined and would provide insight into the conservation of the role of this region throughout the receptor family.
The pharmacology of the P2X receptor also raises some interesting questions about the agonist binding site. If there were a simple agonist binding pocket one might suppose that ADP would have some affinity for the site however at the P2X1 receptor it is ineffective as either an agonist or antagonist (Mahaut-Smith et al. 2000; Vial et al. 2003). These findings suggested that compared to the binding of the adenine ring or ribose the detection/co-ordination of the three phosphate residues plays a dominant role in determining the affinity of drugs at the agonist binding site. It is interesting that longer chain phosphates also activate the channel with for example adenosine tetraphosphate and the diadenosine polyphosphate Ap5A acting as agonists (Lewis et al. 2000). This suggests that the structure of the binding site that recognises the phosphate tail of ATP is relatively open-ended and can accommodate additional substitutions i.e. in the case of Ap5A essentially an ADP molecule bound to the end of ATP. The idea that the binding site can accommodate substitutions at the end of the phosphate tail is also supported by studies shown that the P2X7 receptor can be activated following ADP-ribosylation (Seman et al. 2003). Recent studies have shown that the P2X7 can be post translationally modified by arginine-specific ADP-ribosyltransferases that transfer ADP-ribose from NAD onto arginines 125 and 133 and that it is the ribosylation of arginine 125 that is sufficient to activate the receptor (Adriouch et al. 2007). At P2X7 receptors ADP is ineffective as an agonist (Adriouch et al. 2007). Adriouch et al. (2007) suggest that the ADP-ribosylation site is close to the ATP binding site and the covalent tethering of ADP-ribose (the ribose effectively acting as a linker from the beta phosphate of ADP) to arginine 125 allows the ADP molecule to now bind at the receptor. This covalent binding would overcome the usual low affinity of ADP for the binding site and presumably allow the binding of the phosphates, ribose and adenine to activate the receptor. The arginine residue is in a cysteine rich domain (Fig. 2) that has been suggested to be involved in zinc potentiation of P2X receptors (see later). This work with the P2X7 receptor has provided additional clues as to the molecular structure of the agonist binding site and nearby regions of the receptor.
The functional response of P2X receptors to ATP consists of the initial agonist binding step and then subsequent conformational change associated with the opening of the ion channel (the gating step). In the mutagenesis studies that have been done on P2X receptors macroscopic currents have been recorded and any changes in ATP potency could arise from effects on agonist binding and/or gating (Colquhoun 1998). A crystal structure would be needed to definitively prove the molecular basis of the ATP binding site. However the balance of current evidence and opinion is strongest that residues equivalent to K68 and K309 (P2X1 receptor numbering, that give the largest decreases in ATP potency) are involved in ATP binding (for example it would be unusual to have an ATP binding protein that did not have positively charged residues involved in agonist binding). Recent studies on mutants of the P2X2 receptor have suggested that K308 also contributes to gating of this channel (Cao et al. 2007). For some of the P2X receptor mutants it is likely that the small changes ATP potency result from modification of the structure of the receptor or the conformational gating changes. It is interesting that if the proposed model of the ATP binding site is correct that the agonist binds at some distance from the regions of the receptor that form the ion conducting pore as seen for cys loop and glutamate ion channels. For example residue K309 is ∼20 amino acids away from the start of the second transmembrane domain. Studies on this possible linker region have suggested that it can play a role in the transduction of agonist binding to channel gating (Yan et al. 2006; Roberts and Evans 2007) and there is evidence from changes in the pattern of MTSEA-biotinylation of introduced cysteine residues that this region undergoes conformational changes on ATP binding (Roberts and Evans 2007).
Overall these studies support a model where ATP action is co-ordinated by positively charged lysine residues interacting with the negatively charged phosphate tail of ATP and it seems likely the adenine ring is co-ordinated by aromatic phenylalanine residues. Whether there are residues that interact directly with the ribose moiety remain to be determined. The mutagenesis studies suggest that P2X receptors share a broadly similar agonist binding pocket, however there are likely to be some variations in the local binding environment or gating movements to account for the differences in agonist sensitivity between receptor subunits.
Regulatory sites
In addition to the orthosteric agonist binding site many receptors have additional allosteric binding sites that may regulate the activity of the receptor. For example the affinity of GABA binding to GABAA receptors can be increased by benzodiazepine binding to a distinct allosteric site. It is now clear that the potency of ATP at P2X receptors may also be regulated by similar allosteric mechanisms. For example sub-maximal P2X receptor currents recorded from nodose, sympathetic and spinal cord neurons (Cloues et al. 1993; Li et al. 1993) were potentiated by low concentrations (<10 μM) of zinc. The effect was due to a left shift in the concentration response to ATP with no change in the maximal response (Cloues et al. 1993; Li et al. 1993), these characteristics and single channel analysis showing that zinc increased opening frequency and burst duration (Cloues 1995) are consistent with low concentrations of zinc acting as an allosteric modulator. Subsequent studies addressed the subtype sensitivity to zinc modulation and showed that P2X2 and P2X4 receptor currents could be increased by low micromolar concentrations of zinc (Brake et al. 1994; Seguela et al. 1996; Soto et al. 1996; Xiong et al. 1999) while P2X1 (Wildman et al. 1999) and P2X7 receptor currents were decreased (Virginio et al. 1997). Further studies have shown that P2X receptor currents can be regulated by other ionic species including copper (Xiong et al. 1999) as well as protons (King et al. 1996; Li et al. 1996; Stoop et al. 1997; Wildman et al. 1998, 1999) and the sensitivity to ionic regulation can be used, in part, to assign molecular P2X receptor subtypes to native receptors (Wildman et al. 1999; North and Surprenant 2000).
The molecular basis of the sites of regulation by metals and pH has been investigated in a number of mutagenesis studies on P2X receptor subtypes. Histidine residues were considered most likely to be involved in these effects as the sensitivity of charge at this residue to pH makes it a candidate for regulation by protons and histidines have also been found as part of the metal binding pockets for both copper and zinc (Richardson et al. 1975; Aitken 1999; Vallee et al. 1999). However the lack of conserved histidine residues in the extracellular domain of the P2X receptor family suggested that there was not a common site of regulation. Analysis of mutagenesis studies on a range of P2X receptors provides an emerging picture of the molecular basis of allosteric sites (Table 1).
P2X2 receptors
P2X2 receptor currents can be potentiated by pH and low micromolar concentrations of zinc by an apparent allosteric effect. Alanine point mutations of seven of the conserved ten cysteine residues in the extracellular domain almost abolished the positive allosteric effects of zinc and indicate that these residues play an important role in zinc action. These mutants also reduced ATP potency at the receptor and it was suggested that the cystiene residues may form disulphide bonds and that the breaking of these bonds resulted in structural changes that can account for the reduced effects of zinc (Clyne et al. 2002a). The suggestion that the cysteine residues may form disulphide bonds was also supported by work on P2X1 receptors (Ennion and Evans 2002). At the P2X1 receptor MTSEA-biotin does not label wild type P2X1 receptors showing that all the cystiene residues are disulphide-bonded or inaccessible. Mutation of cysteines 126, 132, 149, 159, 217 or 227 resulted in labelling of the receptor with MTSEA-biotin indicating the exposure of a free cysteine residue created by the disruption of a disulphide bond and this work provides biochemical evidence to suggest at least three disulphide bonds (Ennion and Evans 2002). These results indicate that the disulphide bonding may be essential for the formation of a metal binding pocket (Clyne et al. 2002a; Ennion and Evans 2002) however direct confirmation for the predicted disulphide bonds remains to be provided.
Histidine residues have been shown to contribute to allosteric binding sites in the P2X2 receptor. Potentiation of P2X2 responses by low micromolar zinc was abolished for either of the two mutants H120A and H213A (Clyne et al. 2002b; Lorca et al. 2005) (however the inhibitory effects of higher concentrations zinc were preserved for these mutants demonstrating the complex nature of zinc action). In subsequent studies cysteine mutants of H120 and H213 were generated and shown to form a disulphide bond between adjacent subunits (Nagaya et al. 2005). This was a very important finding and provided not only evidence that the zinc binding site forms at the interface between subunits but also some molecular distances within the P2X receptor. It is also interesting to note that these histidine residues are close to conserved cysteine residues and as suggested above disulphide bonds could stabilise the zinc binding site (Fig. 2). Using chimeras the study also showed that the level of zinc potentiation was dependent on the number of functional intersubunit zinc binding sites (Nagaya et al. 2005). Subsequent work has suggested the region around the zinc binding site is moderately flexible (Tittle et al. 2007).
A distinct histidine residue has also been shown to contribute to the pH sensitivity of the P2X2 receptor. The mutant H319A results in a decrease in pH sensitivity of the receptor (with no effect on zinc sensitivity) and in contrast to WT receptors the peak current was reduced when the pH was changed from pH 7.5 to 6.5 (Clyne et al. 2002b). The contribution of H319 as a proton sensor was also tested by making the H319K mutation to mimic the effects of protonation of the histidine. The ATP potency of the H319K mutant was increased ∼40-fold consistent with protonation of H319 playing a central role in pH regulation of the channel (Clyne et al. 2002b). It is interesting that this residue is in a region that has been suggested to be part of a linker region connecting the ligand binding domain to the channel pore (Yan et al. 2006; Roberts and Evans 2007) and protonation of H319 could play a role in regulation of gating of the channel that could account for the positive allosteric effect on ATP potency.
P2X3 receptors
P2X3 receptors are sensitive to pH with acidification reducing the amplitude of evoked currents at an EC50 concentration of agonist (Stoop et al. 1997). Acidification also leads to a slowing in the time-course of P2X3 receptor currents and resulted in a depression of currents evoked by low concentrations of agonist but facilitated currents in response to high agonist concentrations and decreased ATP potency (Gerevich et al. 2007). Interestingly acidification also shortened the time required for the P2X3 receptor to recover from desensitisation with the time for 50% recovery from the fully desensitised state speeding from ∼1 min at pH 7.4 to ∼25 s at pH 5.8. (Gerevich et al. 2007). The mutant H206A abolished the pH dependent effect on the maximum response as well as the time course and reduced the decrease in agonist potency from pH 7.4 to 5.8 from ∼4-fold to ∼2-fold. This residue in the P2X3 receptor is close (5–10 residues) to the conserved FT region (185–186 P2X1 receptor numbering) that has been suggested to be involved in ATP action (Roberts et al. 2006) and also K190 that reduced ATP potency at both P2X1 and more dramatically at P2X2 receptors (Ennion et al. 2000; Jiang et al. 2000). The mutation in the first transmembrane domain H45A had no effect on the potentiation of the maximum response but produced a greater decrease in ATP potency (Gerevich et al. 2007). Previous studies have shown that mutations within the transmembrane domains can change agonist potency most likely through an effect on channel gating.
P2X4 receptors
At rat PX4 receptors 10 μM zinc produced a modest ∼3-fold increase in ATP potency at the receptor with no effect on the maximum response to ATP or the Hill coefficient (Acuna-Castillo et al. 2000). In contrast copper (300 μM) had no effect on the ATP sensitivity but reduced responses by ∼60% (Acuna-Castillo et al. 2000). The effects of zinc were instantaneous whereas those of copper took time to develop. Site directed mutagenesis has been used to investigate the molecular basis of the action of copper and zinc at the P2X4 receptor. These studies have shown that alanine mutation of either H140 or D138 abolished inhibition by copper (Coddou et al. 2003, 2007). Interestingly these alanine mutations also increased the potentiation by zinc by 2–4-fold. These results suggest there may be some overlap in the residues that mediate the actions of zinc and copper (or that zinc may have sites that mediate both positive and negative effects—although it is clear that they are close together and associated with the cysteine rich region, Fig. 2). In addition alanine mutation of D129 reduced the extent of copper inhibition but had no effect on the effects of zinc (Coddou et al. 2007).
Recent work on the P2X4 receptor identified residues associated with zinc potentiation at the receptor. The facilitatory effects of zinc were abolished for the C132A mutant (but not C126A) and significantly reduced for the mutant T133A, however neither of these mutants had an effect on the inhibition by copper (Coddou et al. 2007). In additional experiments they showed that MTSET that had no effect on ATP responses but reduced by ∼50% the potentiation by zinc, and the authors suggest that this results from modification of C132. Taking these results together Coddou et al. suggest that the C132 residue is not exclusively involved in disulphide bonding but that it contributes to the zinc binding site. For the P2X1 and P2X2 receptors it has been suggested that the cysteine residue 132 forms a disulphide bond with residue cysteine 159 (both P2X1 and P2X4 numbering) (Clyne et al. 2002a; Ennion and Evans 2002). At P2X2 receptors alanine mutation of seven of the conserved cysteine residues (including the equivalents of C126 and C132 in the P2X4 receptor) reduced zinc potentiation and it was suggested these effects resulted from structural alterations in the receptor due to the breaking of disulphide bonds (see above). This raises the possibility that the C132A mutation in the P2X4 receptor leads to the breaking of a disulphide bond (predicted with C159, and the effects of the C159A mutant were not tested) and this changes the structure of the protein and removes the zinc modulatory site. Support for the free cysteine at position 132 in the P2X4 receptor was also suggested from the effects of MTSET on zinc potentiation (Coddou et al. 2007). At P2X2 receptors that are potentiated by zinc MTSET has no effect (Clyne et al. 2002a) on WT channels. However MTSET reduced zinc potentiation at cysteine mutants of H120 and H213 that have been shown to be involved in zinc regulation at the P2X2 receptor (Nagaya et al. 2005). This suggests that MTSET can access and partially block the zinc binding site of P2X2 receptors and suggests that any cysteine residues in the WT receptor are unavailable for modification either as they are part of a disulphide bond or are deep in the zinc binding site. MTSET can also permeate through P2X receptor channels (Rassendren et al. 1997) and blocks currents at P2X2 receptors with cysteine substitution mutations at positions D15 and P19 (Jiang et al. 2001). Splice variants of P2X receptors and mutational studies have indicated that the intracellular domains of the receptor can regulate the gating of P2X receptor channels. There are a number of cysteine residues in the intracellular domains of the P2X4 receptor and this raises the possibility that these residues could contribute to the regulatory effects of MTSET. However what is clear from these studies is that the cysteine rich region plays a role in zinc regulation.
P2X4 receptors are also sensitive to regulation by changes in pH with acidification inhibiting ATP evoked responses (Stoop et al. 1997; Clarke et al. 2000). At the rat P2X4 receptor the histidine 286 to alanine mutation had no effect on regulation by copper or zinc (Coddou et al. 2003) however in the human receptor it abolished the sensitivity to protons (Clarke et al. 2000). Interestingly this is close to the NFR motif that we have shown to be involved in ATP action/binding at the receptor.
P2X7 receptors
Agonist evoked P2X7 receptor currents are inhibited by both zinc and copper (Virginio et al. 1997). The site(s) of action of zinc and copper have been determined in mutagenesis studies (Acuna-Castillo et al. 2007; Liu et al. 2008); these groups used the rat P2X7 receptor and looked at the effects of mutation of histidine, glutamate, aspartate and lysine residues. However the effects of some of the mutations appeared to be dependent on the expression system or the agonist used. When expressed in Xenopus oocytes (Acuna-Castillo et al. 2007) ATP evoked currents at the mutant H267A was unaffected by copper and zinc, H201A and H130A had reduced sensitivity to copper and H219A was less sensitive to zinc inhibition. H201A and H130A were also shown to be involved in magnesisum inhibition of P2X7 receptor currents (Acuna-Castillo et al. 2007). In studies with HEK293 cells a different pattern was seen on ATP evoked responses (Liu et al. 2008), H62A and D197A reduced inhibition by copper or zinc and the double mutant H62AD197A abolished the inhibitory effects of these metals [the H62A mutant showed no change in sensitivity to copper or zinc compared to wild type in oocyte studies and the reason for the difference in sensitivity between the expression systems remains to be determined (Acuna-Castillo et al. 2007)]. H201A, and H267A had a small effect on copper or zinc inhibition (however when BzATP was used as the agonist the inhibitory effects of copper and zinc at these mutants and wild type P2X7 receptors were indistinguishable) (Liu et al. 2008). The consensus for ATP action is that H201A and H267A can be involved in regulation by copper although the relative effect of these mutation appears to be dependent on the expression system.
The differential sensitivity of P2X receptors to regulation by zinc and pH is supported by the mutagenesis studies summarised above that show that there is no consensus in the amino acids involved in regulation and this is consistent with the lack of conservation of residues at these positions. For residues suggested to be involved in pH regulation of P2X2,3,4,7 receptors there appears to be no pattern emerging from the different subunits and this suggests that pH regulation may not have a common basis and be subunit specific (Table 1). However a pattern appears to emerge when considering some of the residues associated with zinc sensitivity where several residues identified from P2X2 and P2X4 receptors appear to be clustered around the cysteine rich region corresponding to the first three conserved cysteine residues (Fig. 2). This cystiene rich region includes the lysine residue that has been shown to be ADP ribosylated in P2X7 receptors and has been suggested to be close to the ATP binding site (Adriouch et al. 2007 see above). This provides some evidence that the zinc allosteric site and the ATP binding site may be in close proximity as had been postulated previously (Tittle et al. 2007). Studies on the P2X2 receptor have shown that it is likely that the residues H120 and H213 from adjacent subunits are close enough together that when they are substituted for cysteine they can form a disulphide bond (Nagaya et al. 2005). This suggests that there is a cysteine rich region (formed from two adjacent subuntis) associated with zinc regulation and is consistent with studies on the P2X2 receptor that showed that zinc potentiation was almost abolished when any of the first seven cysteine residues was mutated to alanine. It is interesting that in the Dictyostelium P2X receptor only cysteine residues corresponding to cysteines 1 and 6 in the mammalian P2X receptors are conserved and the cysteine rich region of ∼46 residues that contains cysteines 2, 3, 4 and 5 as well as residues involved in zinc potentiation is absent. However from the prediction based on mutagenesis studies of P2X1 and P2X2 receptors the conserved cysteines 1 and 6 of mammalian receptors that are predicted to form a disulphide bond are present (and correspond to cysteines 3 and 4 of the Dd P2X receptor), and could form a disulphide bond to stabilise the Dictyostelium receptor.
Summary
Mutagenesis studies have clearly highlighted a number of amino acids that play a key role in regulating ATP sensitivity and allosteric regulation by zinc and pH. Recent studies have suggested that the ATP binding site and the cysteine rich zinc regulatory site are close (Fig. 2). The introduction of cysteine residues and the formation of disulphide bonds suggests that both the ATP binding site and the zinc allosteric site are formed at the interface between subunits. The question that remains to be determined is which part of the ATP site and the zinc site are on the same subunit. These mutagenesis studies have given us considerable insight into residues involved in ATP action at P2X receptors and have allowed the development and refinement of models. These studies with disulphide bonding and the ADP ribosylation have given us some molecular distances for the P2X receptor subunits and will be useful for validation of any future crystal structure that is produced of the receptor. In other channels mutagenesis studies have generally been effective in predicting important information on the structure-function that have been supported by subsequent crystal structures. A crystal structure for the P2X receptor is eagerly awaited and it is promising that recently a structure for an acid sensing trimeric ion channel with two transmembrane domains has been produced (Jasti et al. 2007).
References
Acuna-Castillo C, Morales B, Huidobro-Toro JP (2000) Zinc and copper modulate differentially the P2X4 receptor. J Neurochem 74:1529–1537
Acuna-Castillo C, Coddou C, Bull P, Brito J, Huidobro-Toro JP (2007) Differential role of extracellular histidines in copper, zinc, magnesium and proton modulation of the P2X7 purinergic receptor. J Neurochem 101:17–26
Adriouch S, Bannas P, Schwarz N, Fliegert R, Guse AH, Seman M, Haag F, Koch-Nolte F (2007) ADP-ribosylation at R125 gates the P2X7 ion channel by presenting a covalent ligand to its nucleotide binding site. FASEB J (1530–6860 Electronic)
Aitken A (1999) Protein consensus sequence motifs. Mol Biotechnol 12:241–253
Bean BP (1990) ATP-activated channels in rat and bullfrog sensory neurons: concentration dependence and kinetics. J Neurosci 10(1):1–10
Brake AJ, Wagenbach MJ, Julius D (1994) New structural motif for ligand-gated ion channels defined by an ionotropic ATP receptor. Nature 371:519–523
Cao L, Young MT, Broomhead HE, Fountain SJ, North RA (2007) Thr339-to serine substitution in rat P2X2 receptor second transmembrane domain causes constitutive opening and indicates a gating role for lys308. J Neurosci 27:12916–12923
Clarke CE, Benham CD, Bridges A, George AR, Meadows HJ (2000) Mutation of histidine 286 of the human P2X4 purinoceptor removes extracellular pH sensitivity. J Physiol 523:697–703
Cloues R (1995) Properties of ATP-gated channels recorded from rat sympathetic neurons: voltage dependence and regulation by Zn2+ ions. J Neurophys 73(1):312–319
Cloues R, Jones S, Brown DA (1993) Zn2+ potentiates ATP-activated currents in rat sympathetic neurons. Pflugers Archiv 424:152–158
Clyne JD, Wang LF, Hume RI (2002a) Mutational analysis of the conserved cysteines of the rat P2X2 purinoceptor. J Neurosci 22:3873–3880
Clyne JD, LaPointe LD, Hume RI (2002b) The role of histidine residues in modulation of the rat P2X(2) purinoceptor by zinc and pH. J Physiol 539:347–359
Coddou C, Acuna-Castillo C, Bull P, Huidobro-Toro JP (2007) Dissecting the facilitator and inhibitor allosteric metal sites of the P2X4 receptor channel: critical roles of Cys-132 for zinc-potentiation and Asp-138 for copper-inhibition. J Biol Chem 282:36879–36886
Coddou C, Morales B, Gonzalez J, Grauso M, Gordillo F, Bull P, Rassendren F, Huidobro-Toro JP (2003) Histidine 140 plays a key role in the inhibitory modulation of the P2X4 nucleotide receptor by copper but not zinc. J Biol Chem 278:36777–36785
Colquhoun D (1998) Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol 125:924–947
Digby HR, Roberts JA, Sutcliffe MJ, Evans RJ (2005) Contribution of conserved glycine residues to ATP action at human P2X1 receptors:mutagenesis indicates that the glycine at position 250 is important for channel function. J Neurochem 95:1744–1754
Ding S, Sachs F (1999) Single channel properties of P2X2 purinoceptors. J Gen Physiol 113:695–720
Ennion S, Hagan S, Evans RJ (2000) The role of positively charged amino acids in ATP recognition by human P2X1 receptors. J Biol Chem 275:29361–29367
Ennion SJ, Evans RJ (2002) Conserved cysteine residues in the extracellular loop of the human P2X(1) receptor form disulfide bonds and are involved in receptor trafficking to the cell surface. Mol Pharmacol 61:303–311
Ennion SJ, Ritson J, Evans RJ (2001) Conserved negatively charged residues are not required for ATP action at P2X(1) receptors. Biochem Biophys Res Commun 289:700–704
Fountain SJ, Parkinson K, Young MT, Cao L, Thompson CR, North RA (2007) An intracellular P2X receptor required for osmoregulation in Dictyostelium discoideum. Nature 448:200–203
Gerevich Z, Zadori ZS, Koles L, Kopp L, Milius D, Wirkner K, Gyires K, Illes P (2007) Dual effect of acid pH on purinergic P2X3 receptors depends on the histidine-206 residue. J Biol Chem 282:33949–33957
Jasti J, Furukawa H, Gonzales EB, Gouaux E (2007) Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449:316–323
Jiang L-H, Rassendren F, Surprenant A, North RA (2000) Identification of amino acid residues contributing to the ATP binding site of a purinergic P2X receptor. J Biol Chem 275:34190–34196
Jiang L-H, Rassendren F, Spelta V, Surprenant A, North RA (2001) Amino acid residues involved in gating identified in the first membrane-spanning domain of the rat p2X2 receptor. J Biol Chem 276:14902–14908
King BF, Ziganshin LE, Pintor J, Burnstock G (1996) Full sensitivity of P2X2 purinoceptor revealed by changing extracellular pH. Br J Pharmacol 117:1371–1373
Lewis CJ, Gitterman DP, Schluter H, Evans RJ (2000) Effects of diadenosine polyphosphates (ApnAs) and adenosine polyphospho guanosines (ApnGs) on rat mesenteric artery P2X receptor ion channels. Br J Pharmacol 129:124–130
Li C, Peoples RW, Weight FF (1996) Proton potentiation of ATP-gated ion channel responses to ATP and Zn2+ in rat nodose ganglion neurons. J Neurophys 76:3048–3058
Li C, Peoples RW, Li Z, Weight FF (1993) Zn2+ potentiates excitatory action of ATP on mammalian neurons. PNAS 90:8264–8267
Liu X, Surprenant A, Mao HJ, Roger S, Xia R, Bradley H, Jiang LH (2008) Identification of key residues coordinating functional inhibition of P2X7 receptors by zinc and copper. Mol Pharmacol 73:252–259
Lorca RA, Coddou C, Gazitua MC, Bull P, Arredondo C, Huidobro-Toro JP (2005) Extracellular histidine residues identify common structural determinants in the copper/zinc P2X2 receptor modulation. J Neurochem 95:499–512
Mahaut-Smith MP, Ennion SJ, Rolf MG, Evans RJ (2000) ADP is not an agonist at P2X1 receptors: evidence for separate receptors stimulated by ATP and ADP on human platelets. Br J Pharmacol 131:108–114
Marquez-Klaka B, Rettinger J, Bhargava Y, Eisele T, Nicke A (2007) Identification of an intersubunit cross-link between substituted cysteine residues located in the putative ATP binding site of the P2X1 receptor. J Neurosci 27:1456–1466
Nagaya N, Tittle RK, Saar N, Dellal SS, Hume RI (2005) An intersubunit Zn2+ binding site in rat P2X2 receptors. J Biol Chem 280:25982–25993
North RA (2002) Molecular physiology of P2X Receptors. Physiol Rev 82:1013–1067
North RA, Surprenant A (2000) Pharmacology of cloned P2X receptors. Ann Rev Pharmacol Toxicol 40:563–580
Rassendren F, Buell G, Newbolt A, North RA, Surprenant A (1997) Identification of amino acid residues contributing to the pore of a P2X receptor. EMBO J 16:3446–3454
Richardson J, Thomas KA, Rubin BH, Richardson DC (1975) Crystal structure of bovine Cu,Zn superoxide dismutase at 3 A resolution: chain tracing and metal ligands. Proc Natl Acad Sci USA 72:1349–1353
Roberts JA, Evans RJ (2004) ATP Binding at Human P2X1 Receptors: contribution of aromatic and basic amino acids revealed using mutagenesis and partial agonists. J Biol Chem 279:9043–9055
Roberts JA, Evans RJ (2005) Mutagenesis studies of conserved proline residues of human P2X receptors for ATP indicate that proline 272 contributes to channel function. J Neurochem 92:1256–1264
Roberts JA, Evans RJ (2006) Contribution of conserved polar glutamine, asparagine and threonine residues and glycosylation to agonist action at human P2X1 receptors for ATP. J Neurochem 96:843–852
Roberts JA, Evans RJ (2007) Cysteine substitution mutants give structural insight and identify ATP binding and activation sites at P2X receptors. J Neurosci 27:4072–4082
Roberts JA, Vial C, Digby HR, Agboh KC, Wen H, Atterbury-Thomas AE, Evans RJ (2006) Molecular properties of P2X receptors. Pflugers Arch 452:486–500
Seguela P, Haghighi A, Soghomonian J-J, Cooper E (1996) A novel neuronal P2X ATP receptor ion channel with widespread distribution in the brain. J Neurosci 16:448–455
Seman M, Adriouch S, Scheuplein F, Krebs C, Freese D, Glowacki G, Deterre P, Haag F, Koch-Nolte F (2003) NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity 19:571–582
Soto F, Garcia-Guzman M, Gomez-Hernandez JM, Hollmann M, Karschin C, Stuhmer W (1996) P2X4: an ATP-activated ionotropic receptor cloned from rat brain. Proc Natl Acad Sci USA 93:3684–3688
Stojilkovic SS, Tomic M, He ML, Yan Z, Koshimizu TA, Zemkova H (2005) Molecular Dissection of Purinergic P2X Receptor Channels. Ann N Y Acad Sci 1048:116–130
Stoop R, Surprenant A, North RA (1997) Different sensitivities to pH of ATP-indiced currents at four cloned P2X receptors. J Neurophys 78:1837–1840
Tittle RK, Power JM, Hume RI (2007) A histidine scan to probe the flexibility of the rat P2X2 receptor zinc-binding site. J Biol Chem 282:19526–19533
Vallee F, Turner MA, Lindley PL, Howell PL (1999) Crystal structure of an inactive duck delta II crystallin mutant with bound argininosuccinate. Biochemistry 38:2425–2434
Vial C, Roberts JA, Evans RJ (2004) Molecular properties of ATP-gated P2X receptor ion channel. Trends Pharmacol Sci 25:487–493
Vial C, Pitt SJ, Roberts J, Rolf MG, Mahaut-Smith MP, Evans RJ (2003) Lack of evidence for functional ADP-activated human P2X1 receptors supports a role for ATP during hemostasis and thrombosis. Blood 102:3646–3651
Virginio C, Church D, North RA, Surprenant A (1997) Effects of divalent cations, protons and calmidazolium at the rat P2X7 receptor. Neuropharmacology 36:1285–1294
Walker JE, Saraste M, Runswick MJ, Gay NJ (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP requiring enzymes and a common nucleotide binding fold. EMBO J 1:945–951
Wildman SS, King BF, Burnstock G (1998) Zn2+ modulation of ATP-responses at recombinant P2X2 receptros and its dependence on extracellular pH. Br J Pharmacol 123:1214–1220
Wildman SS, King BF, Burnstock G (1999) Modulatory activity of extracellular H+ and Zn2+ on ATP-responses at rP2X1 and rP2X3 receptors. Br J Pharmacol 128:486–492
Wilkinson WJ, Jiang LH, Surprenant A, North RA (2006) Role of Ectodomain Lysines in the Subunits of the Heteromeric P2X2/3 Receptor. Mol Pharmacol 70:1159–1163
Xiong K, Peoples RW, Montgomery JP, Chiang Y, Stewart RR, Weight FF, Li C (1999) Differential modulation by copper and zinc of P2X2 and P2X4 receptor function. J Neurophysiol 81:2088–2094
Yan Z, Liang Z, Obsil T, Stojilkovic SS (2006) Participation of the Lys313-Ile333 sequence of the purinergic P2X4 receptor in agonist binding and transduction of signals to the channel gate. J Biol Chem 281:32649–32659
Yan Z, Liang Z, Tomic M, Obsil T, Stojilkovic SS (2005) Molecular determinants of the agonist binding domain of a P2X receptor channel. Mol Pharmacol 67:1078–1088
Zemkova H, Yan Z, Liang Z, Jelinkova I, Tomic M, Stojilkovic SS (2007) Role of aromatic and charged ectodomain residues in the P2X(4) receptor functions. J Neurochem 102:1139–1150
Acknowledgments
I am grateful for the support of the Wellcome Trust and British Heart Foundation and to Dr. Lin-Hua Jiang for comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Evans, R.J. Orthosteric and allosteric binding sites of P2X receptors. Eur Biophys J 38, 319–327 (2009). https://doi.org/10.1007/s00249-008-0275-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-008-0275-2