
Abstract
Aims/hypothesis
We determined the contribution to insulin resistance of the PH domain leucine-rich repeat protein phosphatase (PHLPP), which dephosphorylates Akt at Ser473, inhibiting its activity. We measured the abundance of PHLPP in fat and skeletal muscle from obese participants. To study the effect of PHLPP on insulin signalling, PHLPP (also known as PHLPP1) was overexpressed in HepG2 and L6 cells.
Methods
Subcutaneous fat samples were obtained from 82 morbidly obese and ten non-obese participants. Skeletal muscle samples were obtained from 12 obese and eight non-obese participants. Quantification of PHLPP-1 in human tissues was performed by immunoblotting. The functional consequences of recombinant PHLPP1 overexpression in hepatoma HepG2 cells and L6 myoblasts were investigated.
Results
Of the 82 obese participants, 31 had normal fasting glucose, 33 impaired fasting glucose and 18 type 2 diabetes. PHLPP-1 abundance was twofold higher in the three obese groups than in non-obese participants (p = 0.004). No differences were observed between obese participants with normal fasting glucose, impaired fasting glucose or type 2 diabetes. PHLPP-1 abundance was correlated with basal Akt Ser473 phosphorylation (r = −0.48; p = 0.001), BMI (r = 0.44; p < 0.0001), insulin (r = 0.35; p < 0.0001) and HOMA (r = 0.38; p < 0.0001). PHLPP-1 abundance was twofold higher in the skeletal muscle of 12 obese participants than in that of eight non-obese participants (p < 0.0001). Insulin treatment of HepG2 cells resulted in a dose- and time-dependent upregulation of PHLPP-1. Overexpression of PHLPP1 in HepG2 cells and L6 myoblasts resulted in impaired insulin signalling involving Akt/glycogen synthase kinase 3, glycogen synthesis and glucose transport.
Conclusions/interpretation
Increased abundance of PHLPP-1, production of which is regulated by insulin, may represent a new molecular defect in insulin-resistant states such as obesity.
Similar content being viewed by others


Introduction
Obesity and type 2 diabetes are serious threats to public health that are reaching epidemic proportions worldwide. Insulin resistance is a key feature of both conditions [1] and is characterised by a reduced ability of target insulin tissues to take up and metabolise glucose in response to insulin. Although some defects in the early steps of the insulin signalling pathway have been reported in insulin target tissues from mouse models, non-human primates and humans [2–14], the underlying molecular mechanisms responsible for impaired insulin signalling have not been completely elucidated and are being intensely investigated. Insulin initiates its action by binding to the insulin receptor and activating its intrinsic tyrosine kinase. This event leads to phosphorylation on tyrosine residues of a variety of docking proteins, including IRS proteins [15, 16]. Once phosphorylated, IRS proteins engage several proteins containing Src homology 2 domains, including the p85 regulatory subunit of phosphatidylinositol 3-kinase. Activated class I phosphatidylinositol 3-kinase generates the lipid phosphatidylinositol 3,4,5-trisphosphate. Increases of the latter lead to activation of a protein kinase cascade, first stimulating the 3-phosphoinositide-dependent protein kinase-1, which phosphorylates the downstream serine/threonine kinase Akt (also known as protein kinase B) on the activation loop (Thr308), followed by phosphorylation on the hydrophobic motif (Ser473) by mammalian target of rapamycin (mTOR), when complexed to rapamycin-insensitive companion of mTOR [17, 18]. Akt plays a pivotal role in mediating metabolic responses induced by insulin, including stimulation of glucose transport, translocation of glucose transporters to the cell surface, inactivation of glycogen synthase kinase (GSK)-3 and stimulation of glycogen synthesis [19, 20]. Some [21–23], but not all [24–27] studies have reported impaired Akt activation in adipose tissue and skeletal muscle in conditions of insulin resistance such as obesity and type 2 diabetes. A possible explanation of these conflicting observations may be related to the fact that three highly homologous Akt isoforms, termed Akt1, Akt2 and Akt3, are produced in mammalian cells as products of distinct genes. The three Akt isoforms have some differential, non-redundant physiological functions. Thus, Akt1 is the most ubiquitous isoform and is mainly involved in growth [28, 29]. Akt2 is produced in insulin-responsive tissues and plays a pivotal role in glucose metabolism [30, 31], whereas Akt3 is essential for brain growth, but not glucose homeostasis [32]. Accordingly, defects in Ser473 phosphorylation and activation of Akt2, but not Akt1 have been reported in skeletal muscle from insulin-resistant morbidly obese and type 2 diabetic participants [33–35], and have been shown to be associated with impaired glucose transport.
Recently, a novel phosphatase, termed PH domain leucine-rich repeat protein phosphatase (PHLPP), was identified, which directly dephosphorylates the hydrophobic motif of Akt (Ser473) resulting in inhibition of kinase activity [36, 37]. PHLPP exists in two isoforms (PHLPP-1 and PHLPP-2), differing for their selectivity in dephosphorylating specific Akt isoforms. Thus PHLPP-1 dephosphorylates Akt2, while PHLPP-2 dephosphorylates Akt1, raising the intriguing possibility that increased abundance of PHLPP-1 may contribute to insulin resistance by specifically affecting Ser473 phosphorylation on Akt2 in insulin-responsive tissues. To test this possibility, we measured the abundance of PHLPP-1 in adipose tissue and skeletal muscle from obese participants with different degrees of glucose tolerance and correlated PHLPP-1 levels with in vivo insulin sensitivity. In addition, we evaluated the functional impact of increased abundance of PHLPP-1 on insulin signalling and glucose metabolism in two insulin-responsive cell lines transfected with human recombinant PHLPP1.
Methods
Study participants
Abdominal subcutaneous adipose tissue samples (1.0–1.5 g) were obtained during laparoscopic adjustable gastric banding (LAGB) from 82 participants with morbid (i.e. grade 3) obesity according to WHO criteria [38]. In 12 of the 82 participants with morbid obesity, tissue samples of rectus abdominis skeletal muscle were obtained during LAGB. Patients with morbid obesity were eligible for LAGB if they fulfilled the following criteria: age 18–66 years; BMI greater than 40 kg/m2 or greater than 35.0 kg/m2 in the presence of co-morbidities [39]; and history of at least two previous attempts to lose weight with dietary and medical measures followed by relapse of obesity. Exclusion criteria were: obesity secondary to endocrinopathies (Cushing’s disease or syndrome, hypothyroidism), gastrointestinal inflammatory diseases, risk of upper gastrointestinal bleeding, pregnancy, alcohol or drug addiction, and previous or current malignancies. Participants were classified as normal fasting glucose (NFG), impaired fasting glucose (IFG) or type 2 diabetes according to the ADA criteria [40]. Insulin sensitivity was estimated by the HOMA index [41].
Abdominal subcutaneous adipose tissue samples were also obtained during elective abdominal surgery from ten non-obese, non-diabetic participants admitted to the hospital for cholecystectomy or total hysterectomy. In eight of these ten non-obese, non-diabetic participants, rectus abdominis skeletal muscle samples were also obtained. Tissue samples were cleaned of all connective tissue and blood, immediately frozen in liquid nitrogen and stored at −70°C until use [39].
All studies were approved by institutional ethics committees and informed written consent was obtained from each participant in accordance with principles of the Declaration of Helsinki.
Antibodies
An anti-PHLPP-1 peptide antibody was generated by immunising rabbits with a mixture of three peptides corresponding respectively to residues 145–157, 623–637 and 1187–1201 of human PHLPP-1. Anti-PHLPP-2 was purchased from Bethyl Laboratories, Montgomery, TX, USA. Anti-phospho-Akt (Thr308), anti-phospho-Akt (Ser473), anti-total Akt, anti-phospho-GSK-3α/β (Ser9/21) and anti-GSK-3α/β were from Cell Signal Technology (Danvers, MA, USA). Anti-actin was from Sigma-Aldrich (St Louis, MO, USA) and anti-tubulin antibody was from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Quantification of PHLPP-1 and PHLPP-2 in human tissues
Proteins were isolated from adipose and skeletal muscle tissues using T-PER buffer (Pierce Biotechnology, Rockford, IL, USA) according to manufacturer’s instructions. Protein concentration was determined with the Bradford assay (DC Protein Assay; Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Equal amounts of proteins were resolved by SDS-PAGE and electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes (Amersham Biosciences, Piscataway, NJ, USA). The membranes were blotted with anti-PHLPP-1, anti-PHLPP-2 or phospho-Akt (Ser473) antibodies. Loading was normalised using anti-actin antibody. Blots were visualised using peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence (Amersham Biosciences) and band densities were quantified by densitometry. To normalise the blots for protein levels, blots were stripped and re-probed with anti-Akt antibody after immunoblotting with anti-phospho-specific antibody. Akt phosphorylation was calculated as the ratio of phosphorylated to total protein expression.
Cell culture procedures
Human hepatoma HepG2 cells and L6 rat skeletal muscle myoblasts were grown at 37°C in air supplemented with 5% (vol./vol.) CO2 in complete culture medium DMEM (with l-glutamine) supplemented with 10% (vol./vol.) FBS and 1% (vol./vol.) antibiotics. HepG2 cells and L6 myoblasts were seeded into 10 cm dishes and 24 h later (at 70–80% confluence) the complete growth medium was replaced with an appropriate volume of DMEM supplemented with 0.25% (wt/vol.) BSA and used for experiments. For time-course studies, HepG2 cells were treated with 1 × 10−8 mol/l insulin and collected at 0, 12, 24 and 48 h. For dose–response studies, HepG2 cells were treated with insulin (1 × 10−9, 1 × 10−8 and 1 × 10−7 mol/l) for 24 h. Cell lysates and immunoblotting were carried out as previously described [42, 43].
Cell transfection with pcDNA3.1-hPHLPP1
HepG2 and L6 cells were transiently transfected with the pcDNA3.1+ expression vector containing the human PHLPP1 cDNA (GenScript, Piscataway, NJ, USA) or with the empty vector using a reagent (Lipofectamine 2000; Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s protocol. After 48 h, transfected HepG2 and L6 cells were incubated in DMEM supplemented with 0.25% (wt/vol.) BSA in the presence or absence of insulin. Thereafter, cells were lysed and equal amounts of protein were resolved by SDS-PAGE and electrophoretically transferred to PVDF membranes. The membranes were then incubated with anti-PHLPP-1, anti-phospho-Akt (Thr308), anti-phospho-Akt (Ser473) or anti-phosphor-GSK-3α/β (Ser9/21) antibody, followed by incubation with secondary antibody. Loading was normalised using anti-actin antibody. Blots were visualised using peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence. Band densities were quantified by densitometry. To normalise the blots for protein levels, blots were stripped and re-probed with the appropriate primary antibodies after being immunoblotted with anti-phospho-specific antibodies.
Glycogen content assay and 2-deoxy-d-glucose uptake
The accumulation of glycogen in HepG2 cells was determined by glycogen assay kit (BioVision, Mountain View, CA, USA), which measures colorimetrically (optical density at 570 nm) the glycogen content. The cells were transfected as described above and serum-starved for 5 h. They were then stimulated with 1 × 10−7 mol/l insulin for 15 min and treated according to the manufacturer’s protocol. Uptake of 2-deoxy-d-glucose was measured as previously reported [44] using 2-deoxy-[14C]glucose (Perkin Elmer, Waltham, MA, USA).
Statistical analysis
Fasting insulin and HOMA values were natural log-transformed for statistical analysis due to their skewed distribution. Group differences of continuous variables were compared using ANOVA or unpaired Student’s t test, as appropriate. Categorical variables were compared by contingency χ2 test. Phenotypic differences between the four groups were tested after adjusting for age and sex by analysis of covariance (general linear model) with Bonferroni correction for multiple comparisons. Partial correlation coefficients adjusted for age and sex were computed between variables. All tests were two-sided. A value of p < 0.05 was considered statistically significant. All analyses were performed using SPSS software version 16.0 for Windows (SPSS, Chicago, IL, USA).
Results
The clinical characteristics of the four study groups are shown in Table 1. By definition, BMI was lower in the non-obese group than in the three groups of obese participants. Of the 82 obese participants, 31 had NFG, 33 had IFG and 18 had type 2 diabetes. Of the 18 diabetic obese participants, six were being treated with metformin.
The obese participants with type 2 diabetes were older than obese individuals with NFG or IFG. After adjusting for age and sex, the three groups of obese participants had significantly higher levels of fasting insulin and HOMA index of insulin resistance than non-obese participants.
We assessed PHLPP-1 protein levels by western blot in total adipose tissue lysates obtained from the whole cohort of morbidly obese participants, who were stratified according to degree of dysglycaemia, and from the cohort of non-obese participants (Fig. 1). PHLPP-1 abundance was twofold higher in the three obese groups than in non-obese participants (p = 0.004). No differences were observed among obese participants with NFG, IFG or type 2 diabetes. After adjusting for sex and age, PHLPP-1 abundance was positively correlated with BMI (r = 0.44; p < 0.0001), fasting insulin levels (r = 0.35; p < 0.0001) and HOMA index of insulin resistance (r = 0.38; p < 0.0001) (Fig. 2). Results were unchanged when the six participants on metformin were excluded from the analysis. By contrast, PHLPP-2 protein abundance in adipose tissue lysates obtained from 37 morbidly obese participants was not correlated with BMI (r = 0.01; p = 0.97), fasting insulin levels (r = 0.19; p = 0.27) and HOMA index of insulin resistance (r = 0.14; p = 0.41) (data not shown).

Immunoblot and quantification of PHLPP-1 in human adipose tissue. Equal amounts of tissue lysates were resolved by SDS-PAGE and transferred to PVDF membranes. The membranes were then incubated with the anti-PHLPP-1 antibody. Blots were visualised using peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence. Band densities were quantified by densitometry. Loading was normalised using anti-actin antibody. Representative immunoblots of three participants from the four study groups are shown. Bars represent the means ± SD for the four study groups. Non-obese, n = 10; obese with NFG, n = 31; obese with IFG, n = 33; obese with type 2 diabetes (T2DM), n = 18; † p < 0.004 vs non-obese participants. AU, arbitrary units

Abundance of PHLPP-1 in human adipose tissue in relation to (a) BMI, (b) fasting plasma insulin and (c) HOMA index. a r = 0.44, p < 0.0001; (b) r = 0.35, p < 0.0001; (c) r = 0.38, p < 0.0001. AU, arbitrary units; dots, individual participants; diagonal lines, regression lines
Since PHLPP-1 dephosphorylates the hydrophobic motif of Akt Ser473, we determined the phosphorylation state of Akt at Ser473 under basal conditions using phospho-specific antibodies. As shown in Fig. 3, we observed markedly reduced levels of basal Akt Ser473 phosphorylation in obese participants (n = 42) as compared with non-obese participants (n = 8) (Fig. 3a). After adjusting for sex and age, basal Ser473 phosphorylation of Akt was inversely correlated with PHLPP-1 protein abundance (r = −0.48; p = 0.001) (Fig. 3b).

Phosphorylation state of Akt at Ser473 under basal conditions in human adipose tissue. Equal amounts of adipose tissue lysates were resolved by SDS-PAGE and transferred to PVDF membranes. The membranes were then incubated with anti-phospho-Akt (Ser473) antibody. Blots were visualised using peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence. Band densities were quantified by densitometry. To normalise the blots for protein levels, blots were stripped and re-probed with anti-total Akt antibody after immunoblotting with anti-phospho-specific antibody. Akt phosphorylation was calculated as the ratio of phosphorylated to total protein abundance. a Representative immunoblots of three non-obese participants (n = 8) and three obese participants (n = 42), with quantification. Bars represent the means ± SD for both groups. † p = 0.005 vs non-obese participants. b Correlation between basal Akt phosphorylation at Ser473 and PHLPP-1 protein abundance; r = −0.48, p = 0.001. AU, arbitrary units; dots, individual participants; diagonal line, regression line
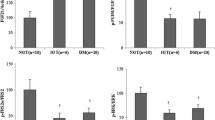
In keeping with data obtained in adipose tissue, PHLPP-1 abundance was twofold higher in the skeletal muscle of 12 obese participants than in that from eight non-obese participants (p < 0.0001) (Fig. 4). Of the 12 obese participants, five had NFG, five IFG and two type 2 diabetes. No differences in PHLPP-1 abundance were observed among obese participants with NFG, IFG or type 2 diabetes (p = 0.54). Basal Ser473 phosphorylation of Akt was markedly reduced in obese participants as compared with non-obese participants (p < 0.0001) (Fig. 4). After adjusting for sex and age, basal Ser473 phosphorylation of Akt in skeletal muscle was inversely correlated with PHLPP-1 protein abundance (r = −0.63; p < 0.0001).

Quantification of PHLPP-1 abundance and of phosphorylation state of Akt at Ser473 under basal conditions in human skeletal muscle. Equal amounts of skeletal muscle lysates were resolved by SDS-PAGE and transferred to PVDF membranes. The membranes were then incubated with anti-PHLPP-1 or anti-phospho-Akt (Ser473) antibody. Blots were visualised using peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence. Band densities were quantified by densitometry. Loading was normalised using anti-tubulin antibody. To normalise the anti-phospho-Akt (Ser473) blots for protein levels, blots were stripped and re-probed with anti-total Akt antibody after immunoblotting with anti-phospho-specific antibody. Akt phosphorylation was calculated as the ratio of phosphorylated to total protein levels. a Representative PHLPP-1 immunoblots from three non-obese participants (n = 8) and three obese participants (n = 12), with quantification. b Representative phospho-Akt immunoblots from three non-obese participants and three obese participants, with quantification. Bars represent the means ± SD for both groups; † p < 0.0001 vs non-obese participants. AU, arbitrary units
As we observed a correlation between PHLPP-1 protein production and insulin levels, we determined whether insulin was able to induce PHLPP-1 production in cultured human hepatoma HepG2 cells. Dose–response and time-course experiments showed that PHLPP-1 protein levels were increased by insulin, with a maximal effect observed at 10−8 mol/l insulin for 24 h (p < 0.05) (Fig. 5a, b).

Immunoblots and quantification of effect of insulin on PHLPP-1 abundance in human HepG2 hepatoma cells. a HepG2 cells were incubated for 24 h with increasing concentrations of insulin or (b) for the indicated periods of time with 100 nmol/l insulin. Cells were lysed, and equal amounts of protein were resolved by SDS-PAGE and transferred to PVDF membranes. The membranes were then incubated with anti-PHLPP-1 antibody. Blots were visualised using peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence and band densities were quantified by densitometry. Loading was normalised using anti-actin (Act) antibody. **p < 0.01 and ***p < 0.001 vs control cells. AU, arbitrary units
To determine the functional impact of PHLPP-1 overproduction in insulin-responsive cell lines, human hepatoma HepG2 cells and L6 rat skeletal muscle cells were transfected with a pcDNA3.1 expression vector containing the human PHLPP1 cDNA or with the empty pcDNA3.1 vector. As shown in Fig. 6a, production of PHLPP-1 in transfected HepG2 cells was threefold higher than in control cells. Insulin-induced phosphorylation of Akt at Ser473 was reduced by ~70% in HepG2 cells transfected with PHLPP1 (Fig. 6b). By contrast, overabundance of PHLPP-1 had no significant effect on the insulin-stimulated phosphorylation of Thr308 (Fig. 6c).

Expression of recombinant human PHLPP1 in human hepatoma HepG2 cells. A pcDNA3.1 expression vector containing human PHLPP1 or an empty pcDNA3.1 expression vector was transiently transfected into HepG2 cells. Transfected HepG2 cells were incubated in the presence or absence of insulin and lysed. Equal amounts of protein were resolved by SDS-PAGE and transferred to PVDF membranes. The membranes were then incubated with anti-PHLPP-1 (a), anti-phospho-Akt (Ser473) antibody (b), anti-phospho-Akt (Thr308) (c) and anti-phospho-GSK-3 (Ser21/9) (d) antibodies. Blots were visualised using peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence. Band densities were quantified by densitometry. Loading was normalised using anti-actin (Act) antibody. To normalise the blots for protein levels, blots were stripped and re-probed with the appropriate primary antibodies after being immunoblotted with anti-phospho-specific antibodies. e Transfected HepG2 cells were incubated in the presence or absence of insulin, and accumulation of glycogen was determined by glycogen assay kit. Bars represent the means ± SD from three independent experiments. **p < 0.01 vs control L6 cells. AU, arbitrary units
GSK-3 has been implicated in the regulation of glycogen synthesis through a mechanism involving its phosphorylation and inactivation by Akt [19]. Insulin-induced GSK-3α/β phosphorylation was decreased by 60% (Fig. 6d) and insulin-stimulated glycogen content was reduced by 65% in HepG2 cells transfected with PHLPP1 as compared with control cells (Fig. 6e). In addition, as shown in Fig. 7a, expression of recombinant PHLPP1 in transfected L6 cells was ~2.8-fold higher than in control cells. Insulin-induced phosphorylation of Akt at Ser473 was markedly reduced in L6 cells transfected with PHLPP1 (Fig. 7b). Moreover, in untransfected L6 cells (not shown) as well as in L6 cells transfected with the empty vector, insulin induced an increase of glucose uptake by twofold, while overexpression of PHLPP1 almost completely abolished the effect of insulin on glucose uptake in these cells (Fig. 7c).

Expression of recombinant human PHLPP1 in rat skeletal muscle L6 cells. A pcDNA3.1 expression vector containing human PHLPP1 or an empty pcDNA3.1 expression vector was transiently transfected into rat skeletal muscle L6 cells. Transfected L6 cells were incubated in the presence or absence of insulin and lysed. Equal amounts of protein were resolved by SDS-PAGE and transferred to PVDF membranes. The membranes were then incubated with anti-phospho-Akt (Ser473) antibody. Blots were visualised using peroxidase-conjugated secondary antibodies followed by enhanced chemiluminescence. Band densities were quantified by densitometry. Loading was normalised using anti-actin (Act) antibody. a PHLPP-1:actin. b To normalise the blots for protein levels, blots were stripped and re-probed with anti-total Akt after being immunoblotted with anti-phospho-specific antibody. c Transfected L6 cells were incubated in the presence or absence of insulin for 15 min and 2-deoxy-[14C]glucose (2-DG) uptake was measured. Bars represent the means ± SD for three independent experiments; **p < 0.01 and ***p < 0.001 vs control L6 cells. AU, arbitrary units
Discussion
The serine/threonine kinase Akt is an important component of the insulin signalling cascade and mediates the majority of the metabolic actions of insulin [17, 18]. Among the three highly homologous Akt isoforms, increasing evidence suggests that Akt2 plays a major role in glucose metabolism [30, 31]. Although conflicting results have been obtained on whether Akt activation in adipose tissue and skeletal muscle is impaired in conditions of insulin resistance [21–27], defects in Ser473 phosphorylation and activation of Akt2, but not of Akt1 have been observed in skeletal muscle of insulin-resistant morbidly obese and type 2 diabetic participants [33–35]. Mechanisms by which Akt is inactivated have, until recently, been poorly documented. Recently, PHLPP-1 was identified as an Akt phosphatase that selectively dephosphorylates Ser473 on Akt2 [36, 37] (electronic supplementary material [ESM] Fig. 1), thus raising the possibility that an increase in PHLPP-1 levels may account for defects in Ser473 phosphorylation of Akt2. We demonstrate here that production of PHLPP-1 is increased in adipose tissue and skeletal muscle of morbidly obese participants as compared with non-obese participants and is inversely related to basal Ser473 phosphorylation of Akt. It has been hypothesised that phosphorylation of the Ser473 residue may not be required for all Akt functions and that some Akt substrates are dependent on Thr308-Ser473, while others may only require Thr308 phosphorylation [45]. Our data suggest that GSK-3, an Akt substrate that regulates key insulin actions in the liver, belongs to the former group, as already suggested by Gao et al. [36]. Importantly, increased abundance of PHLPP-1 was positively correlated with BMI, fasting insulin levels and HOMA index of insulin resistance. Our finding that abundance of PHLPP-1 is not further increased in obese participants with IFG or type 2 diabetes suggests that increased abundance of PHLPP-1 may represent a molecular defect associated with a state of insulin resistance and compensatory hyperinsulinaemia, rather than with hyperglycaemia. This possibility is consistent with our experiments performed with human hepatoma HepG2 cells and showing that exposure to insulin induced an upregulation of PHLPP-1 in a dose- and time-dependent manner.
We did not observe any correlation between PHLPP-2 production in adipose tissue of morbidly obese participants and BMI, fasting insulin levels or HOMA index. These data are consistent with those that have been previously reported in skeletal muscle biopsies from controls and type 2 diabetic participants and show that PHLPP1, but not PHLPP2 mRNA is upregulated in type 2 diabetic individuals in association with a defect in Ser473 phosphorylation of Akt2 [35]. The functional significance of PHLPP-1 production in regulating physiological and pathophysiological responses to metabolic actions of insulin has not yet been examined. To address this issue, we transfected two insulin-responsive cell lines with human recombinant PHLPP1 and assessed its effects on insulin signalling and glucose metabolism. We found that threefold higher levels of PHLPP-1 in human hepatoma HepG2 cells resulted in a marked reduction of insulin-induced phosphorylation of Akt at Ser473, paralleled by a decrease in GSK-3α/β phosphorylation and glycogen synthesis. Similarly, 2.8-fold higher levels of PHLPP-1 in L6 rat skeletal muscle cells resulted in a marked decrease in insulin-stimulated phosphorylation of Akt at Ser473, which was associated with a decrease in glucose transport. Taken together, the present results raise the possibility that changes in abundance of PHLPP-1 may contribute to insulin resistance of obese participants by negatively regulating Akt activity through dephosphorylation of Akt at Ser473.
The strengths of the present study include the large sample size, the inclusion of both sexes, the analysis of obese participants with different degrees of glucose homeostasis, the homogeneity of the sample with detailed clinical characterisation and the transfection experiments carried out in two insulin-responsive cell models. Nevertheless, when interpreting our results, some limitations should be considered. Due to the limited amount of material accessible, it was not possible to immunoprecipitate each Akt isoform to measure the specific phosphorylation pattern directly from tissue samples upon insulin stimulation. In addition, we only evaluated basal Akt phosphorylation, which revealed decreased Ser473 phosphorylation in tissues from obese participants. Moreover, it is unclear whether changes in PHLPP-1 abundance observed in obese participants are due to primary cellular abnormalities or to secondary derangements acting in vivo. However, the finding that PHLPP-1 was not further increased in obese participants with IFG or type 2 diabetes and was upregulated in vitro by insulin exposure suggests that increased production of PHLPP-1 in obesity may be a secondary event caused by hyperinsulinaemia, rather than a primary defect.
In conclusion, we found that PHLPP-1 abundance is increased in adipose tissue and skeletal muscle of obese participants, and is also significantly related to BMI and insulin resistance. In view of these and previous results [35], we hypothesise that changes in abundance of PHLPP-1, the cellular levels of which are increased by insulin exposure, may represent a molecular defect common to insulin-resistant states, including obesity and type 2 diabetes.
Abbreviations
- GSK:
-
Glycogen synthase kinase
- IFG:
-
Impaired fasting glucose
- LAGB:
-
Laparoscopic adjustable gastric banding
- mTOR:
-
Mammalian target of rapamycin
- NFG:
-
Normal fasting glucose
- PHLPP:
-
PH domain leucine-rich repeat protein phosphatase
- PVDF:
-
Polyvinylidene fluoride
References
Sesti G, Federici M, Lauro D, Sbraccia P, Lauro R (2001) Molecular mechanism of insulin resistance in type 2 diabetes mellitus: role of the insulin receptor variant forms. Diabetes Metab Res Rev 17:363–373
Sesti G (2006) Pathophysiology of insulin resistance. Best Pract Res, Clin Endocrinol Metab 20:665–679
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96
Folli F, Saad MJ, Backer JM, Kahn CR (1993) Regulation of phosphatidylinositol 3-kinase activity in liver and muscle of animal models of insulin-resistant and insulin-deficient diabetes mellitus. J Clin Invest 92:1787–1794
Chavez AO, Lopez-Alvarenga JC, Tejero ME et al (2008) Physiological and molecular determinants of insulin action in the baboon. Diabetes 57:899–908
Federici M, Zucaro L, Porzio O et al (1996) Increased expression of insulin/insulin-like growth factor-I hybrid receptors in skeletal muscle of non-insulin-dependent diabetes mellitus subjects. J Clin Invest 98:2887–2893
Caro JF, Sinha MK, Raju SM et al (1987) Insulin receptor kinase in human skeletal muscle from obese subjects with and without noninsulin dependent diabetes. J Clin Invest 79:1330–1337
Arner P, Pollare T, Lithell H, Livingston JN (1987) Defective insulin receptor tyrosine kinase in human skeletal muscle in obesity and type II (non-insulin-dependent) diabetes mellitus. Diabetologia 30:437–440
Maegawa H, Shigeta Y, Egawa K, Kobayashi M (1991) Impaired autophosphorylation of insulin receptors from abdominal skeletal muscles in nonobese subjects with NIDDM. Diabetes 40:815–819
Nolan JJ, Freidenberg G, Henry R, Reichart D, Olefsky JM (1994) Role of human skeletal muscle insulin receptor kinase in the in vivo insulin resistance of noninsulin-dependent diabetes mellitus and obesity. J Clin Endocrinol Metab 78:471–477
Sinha MK, Pories WJ, Flickinger EG, Meelheim D, Caro JF (1987) Insulin-receptor kinase activity of adipose tissue from morbidly obese humans with and without NIDDM. Diabetes 36:620–625
Goodyear LJ, Giorgino F, Sherman LA, Carey J, Smith RJ, Dohm GL (1995) Insulin receptor phosphorylation, insulin receptor substrate-1 phosphorylation, and phosphatidylinositol 3-kinase activity are decreased in intact skeletal muscle strips from obese subjects. J Clin Invest 95:2195–2204
Björnholm M, Kawano Y, Lehtihet M, Zierath JR (1997) Insulin receptor substrate-1 phosphorylation and phosphatidylinositol 3-kinase activity are decreased in skeletal muscle from NIDDM subjects following in vivo insulin stimulation. Diabetes 46:524–527
Sesti G, Federici M, Hribal ML, Lauro D, Sbraccia P, Lauro R (2001) Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. FASEB J 15:2099–2111
Folli F, Saad MJ, Backer JM, Kahn CR (1992) Insulin stimulation of phosphatidylinositol 3-kinase activity and association with insulin receptor substrate 1 in liver and muscle of the intact rat. J Biol Chem 267:22171–22177
Giovannone B, Scaldaferri ML, Federici M et al (2000) Insulin receptor substrate (IRS) transduction system: distinct and overlapping signaling potential. Diabetes Metab Res Rev 16:434–441
Burgering BMT, Coffer PJ (1995) Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 376:599–602
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789
Kohn AD, Summers SA, Birnbaum MJ, Roth RA (1996) Expression of a constitutively active Akt Ser/Thr kinase in 3T3–L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem 271:31372–31378
Rondinone CM, Carvalho E, Wesslau C, Smith U (1999) Impaired glucose transport and protein kinase B activation by insulin, but not okadaic acid, in adipocytes from subjects with type II diabetes mellitus. Diabetologia 42:819–825
Carvalho E, Eliasson B, Wesslau C, Smith U (2000) Impaired phosphorylation and insulin-stimulated translocation to the plasma membrane of protein kinase B/Akt in adipocytes from type II diabetic subjects. Diabetologia 43:1107–1115
Krook A, Roth RA, Jiang XJ, Zierath JR, Wallberg-Henriksen H (1998) Insulin stimulated Akt kinase activity is reduced in skeletal muscle from NIDDM subjects. Diabetes 47:1281–1286
Beeson M, Sajan MP, Dizon M et al (2003) Activation of protein kinase C-ζ by insulin and phosphatidylinositol-3,4,5-(PO4)3 is defective in muscle in type 2 diabetes and impaired glucose tolerance. Amelioration by rosiglitazone and exercise. Diabetes 52:1926–1934
Kim Y-B, Nikoulina SE, Ciaraldi TP, Henry RR, Kahn BB (1999) Normal insulin dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J Clin Invest 104:733–741
Kim YB, Kotani K, Ciaraldi TP, Henry RR, Kahn BB (2003) Insulin-stimulated PKC-ζ/λ activity is reduced and PDK-1 activity is normal in muscle of insulin resistant humans. Diabetes 52:1935–1942
Sajan MP, Standaert ML, Miura A et al (2004) Impaired activation of protein kinase C-ζ by insulin and phosphatidylinositol-3,4,5-(PO4)3 in cultured preadipocyte-derived adipocytes and myotubes of obese subjects. J Clin Endocrinol Metab 89:3994–3998
Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ (2001) Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem 276:38349–38352
Chen WS, Xu PZ, Gottlob K et al (2001) Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev 15:2203–2208
Cho H, Mu J, Kim JK et al (2001) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292:1728–1731
Bouzakri K, Zachrisson A, Al-Khalili L et al (2006) siRNA-based gene silencing reveals specialized roles of IRS-1/Akt2 and IRS-2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metab 4:89–96
Tschopp O, Yang ZZ, Brodbeck D et al (2005) Essential role of protein kinase B gamma (PKB gamma/Akt3) in post natal brain development but not in glucose homeostasis. Development 132:2943–2954
Brozinick JT Jr, Roberts BR, Dohm GL (2003) Defective signaling through Akt-2 and -3 but not Akt-1 in insulin-resistant human skeletal muscle: potential role in insulin resistance. Diabetes 52:935–941
Gosmanov AR, Umpierrez GE, Karabell AH, Cuervo R, Thomason DB (2004) Impaired expression and insulin-stimulated phosphorylation of Akt-2 in muscle of obese patients with atypical diabetes. Am J Physiol Endocrinol Metab 287:E8–E15
Cozzone D, Fröjdö S, Disse E et al (2008) Isoform-specific defects of insulin stimulation of Akt/protein kinase B (PKB) in skeletal muscle cells from type 2 diabetic patients. Diabetologia 51:512–521
Gao T, Furnari F, Newton AC (2005) PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell 18:13–24
Brognard J, Sierecki E, Gao T, Newton AC (2007) PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell 25:917–931
WHO Expert Committee (1995) Physical status: the use and interpretation of anthropometry: report of a WHO expert committee. WHO Tech Rep Ser 854:1–452
Pontiroli EA, Pizzocri P, Librenti MC et al (2003) Laparoscopic adjustable gastric banding for the treatment of morbid (grade 3) obesity and its metabolic complications: a three-year study. J Clin Endocrinol Metab 87:3555–3561
American Diabetes Association (2010) Diagnosis and classification of diabetes mellitus. Diabetes Care 33:S62–S69
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985) Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Federici M, Porzio O, Lauro D et al (1998) Increased abundance of insulin/insulin-like growth factor-I hybrid receptors in skeletal muscle of obese subjects is correlated with in vivo insulin sensitivity. J Clin Endocrinol Metab 83:2911–2915
Federici M, Porzio O, Zucaro L et al (1997) Increased abundance of insulin/IGF-I hybrid receptors in adipose tissue from NIDDM patients. Mol Cell Endocrinol 135:41–47
Raciti GA, Iadicicco C, Ulianich L et al (2010) Glucosamine-induced endoplasmic reticulum stress affects GLUT4 expression via activating transcription factor 6 in rat and human skeletal muscle cells. Diabetologia 53:955–965
Mendoza MC, Blenis J (2007) PHLPPing it off: phosphatases get in the Akt. Mol Cell 25:798–800
Acknowledgements
This work was supported by the following grants: EFSD/Novo Nordisk Programme in Diabetes Research to M. L. Hribal; Italian Ministry of Health RF2007-631176 and Italian Ministry of University 200775N24E-003 to G. Sesti.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Schematic representation of the mechanism of action of PHLPP-1 in the insulin signalling cascade. PI 3-kinase, phosphatidylinositol 3-kinase (PDF 230 kb)
Rights and permissions
About this article
Cite this article
Andreozzi, F., Procopio, C., Greco, A. et al. Increased levels of the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP)-1 in obese participants are associated with insulin resistance. Diabetologia 54, 1879–1887 (2011). https://doi.org/10.1007/s00125-011-2116-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2116-6